Меню
Меню





 Все темы
Все темы
Атрофия нейронов при болезни Кеннеди запускается всплеском тестостерона сразу после рождения
Бульбоспинальная мышечная атрофия (болезнь Кеннеди) — редкое генетическое нейродегенеративное заболевание, вызванное экспансией CAG-повторов в гене AR; проявляется обычно у взрослых мужчин. Исследователи из Японии и США обнаружили, что естественный выброс тестостерона сразу после рождения вызывает чрезмерную активацию мутантного AR в моторных нейронах у новорожденных мышей, моделирующих болезнь. В результате повышается доля изоформы RESR4 транскрипционного репрессора REST, которая, наоборот, активирует экспрессию целевых генов. Это приводит к гиперактивации нервных клеток, а в конечном счете — к их гибели. Введение при рождении антисмысловых олигонуклеотидов, таргетирующих мутантный AR или REST4, значительно подавляло процесс разрушения нейронов. Оба метода лечения улучшали выживаемость и двигательные функции у мышей в возрасте 13 недель.
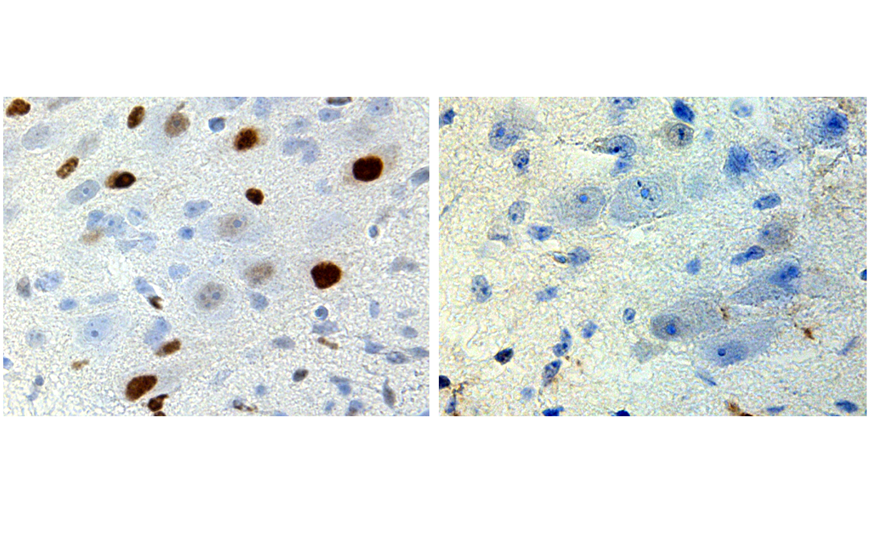
Credit:
Hirunagi et al., 2026 | Пресс-релиз
Бульбоспинальная мышечная атрофия (болезнь Кеннеди, БК) — редкое генетическое X-сцепленное нейродегенеративное заболевание, вызванное экспансией CAG-повторов в гене AR, кодирующих полиглутамин. Проявляется обычно у взрослых мужчин. Андрогеновый рецептор (AR) с полиглутаминовым участком накапливается в ядре. Транскрипция и основные функции клеток нарушаются, что в конечном итоге приводит к дегенерации как двигательных нейронов, так и скелетных мышц.
Ранее на мышиных моделях БК была показана эффективность антисмысловых олигонуклеотидов, таргетирующих AR, при введении до появления симптомов (5 или 8–11 недель). Но есть все больше свидетельств того, что аномалии в центральной нервной системе проявляются еще на более ранних этапах развития. Этой проблемой занялись исследователи из Японии и США.
Опыты проводили на модельных мышах AR-97Q, экспрессирующих человеческий AR с 97 CAG-повторами. Мышечная слабость у них развивается в возрасте 8–9 недель, за чем следует мышечная атрофия и потеря веса в возрасте 13–15 недель.
У самцов мутантный AR в ядрах двигательных нейронов и клетках скелетных мышц накапливаются уже через день после рождения. Если ввести самкам в этом возрасте 20 мкг тестостерона, то у них AR тоже начинает накапливаться в ядрах. Однако формирования белковых агрегатов на ранних стадиях не происходит.
Авторы предположили, что накопление AR с полиглутаминовым участком в ядре влияет на развитие двигательных нейронов на ранних постнатальных стадиях. Они внутрижелудочково вводили антисмысловые олигонуклеотиды, подавляющего экспрессию человеческого AR и мышиного Ar, в первый день после рождения. Снижение уровня экспрессии AR было временным и ограничивалось центральной нервной системой (уровень не снижался в скелетных мышцах).
Ведение антисмысловых олигонуклеотидов существенно продлевало жизнь мышам, тормозило развитие мышечной слабости и атрофию нейронов. Эффект зависел от дозы. Антисмысловой олигонуклеотид не повлиял на уровни олигомерных и мономерных AR в возрасте 13 недель.
Напротив, введение тестостерона через день после рождения снижало выживаемость, усугубляло двигательную дисфункцию и потерю веса у самцов AR-97Q, но не дикого типа. На выживаемость самок AR-97Q тестостерон не влиял, но двигались те хуже, чем самки дикого типа.
Почему мутантный AR токсичен для нейронов? РНК-секвенирование спинного мозга на седьмой день после рождения показало значительное изменение транскриптома у самцов AR-97Q. У них активнее экспрессировался транскрипционный репрессор REST. Но целевые гены REST, включая гены глутаматергических синапсов, активировались в двигательных нейронах у мышей AR-97Q. При этом наличие мутантного AR подавляло постнатальное созревание двигательных нейронов, а введение тестостерона вскоре после рождения усугубляло ситуацию. Это приводило к повышенной возбудимости двигательных нейронов еще до развития симптомов.
Почему повышение экспрессии REST усиливает экспрессию синаптических генов? Оказалось, что в спинном мозге мышей AR-97Q на седьмой день после рождения был повышен уровень экспрессии Rest4 — специфичной для нейронов изоформы, которая подавляет ингибирующий эффект REST. По-видимому, мутантный AR влияет на сплайсинг REST и способствует росту соотношения REST4 к REST в двигательных нейронах. В результате роль REST как репрессора подавляется, а его таргетные гены активируются на более высоком уровне.
Антисмысловой олигонуклеотид, направленный на Rest4, подавлял как его экспрессию, так и экспрессию целевых генов REST. В результате нейроны престают быть такими гиперчувствительными, мыши показывают лучшие результаты при проверке баланса и координации, а также мышечной силы (незначительно).
«Пожалуй, самым примечательным открытием стало то, что препарат, введенный при рождении для воздействия на мутантный белок, продолжал защищать двигательные нейроны спустя месяцы. И это несмотря на то, что действие препарата прекратилось в течение двух недель. Это говорит о том, что вмешательство в нужный момент на ранних этапах жизни может иметь долгосрочные последствия, спустя долгое время после прекращения лечения», — сказал профессор Томоки Хирунаги из Высшей медицинской школы Нагойского университета.
Нейродегенерация при болезни Ниманна — Пика типа С вызвана чрезмерной активацией микроглии
Источник:
Tomoki Hirunagi, et al. Restoring early postnatal synaptic dysregulation rescues motor neuron degeneration in a mouse model of Spinal and Bulbar Muscular Atrophy // Nature Communications (2026), published 27 March 2026, DOI: 10.1038/s41467-026-70244-2
Вам будет интересно

 43
43
 0
0


 36
0
36
0