Меню
Меню





 Все темы
Все темы
МД-2025: Молекулярная диагностика наследственных заболеваний — свежие вызовы, новые решения
Как оптимизировать алгоритм селективного скрининга наследственных коагулопатий? Почему классическое понятие моногенной болезни начинает устаревать? Насколько сильно мы недооцениваем распространенность болезней экспансии и что поможет их выявить? Как часто носитель рецессивной патогенной мутации может стать единственной причиной заболевания у своего ребенка? Ответы на эти и многие другие вопросы прозвучали на секции, посвященной молекулярной диагностике моногенных заболеваний.

Андрей Константинов
Секцию открыл доклад о диагностике моногенных форм коагулопатий, который прочитал Александр Пушков, к.б.н, ФГАУ «НМИЦ здоровья детей».
Наилучшим инструментом, который придумала современная медицинская наука для выявления наследственных патологий, является массовый скрининг новорожденных. Второй способ — селективный скрининг, который проводится с определенными критериями отбора.
Заболевания, на которые проводится селективный скрининг, должны встречаться с частотой не менее 1:150 000 и без ранней диагностики и лечения приводить к серьезному снижению жизнеспособности и инвалидизации. Для направления на скрининг само заболевание должно быть заподозрено в каком-то виде специалистом медико-генетического профиля. Отобранных пациентов направляют на первичный скрининг (как правило, биохимическими методами), а затем проводят подтверждающую молекулярную диагностику.
Сегодня в фокусе диагностика редких форм коагулопатий, то есть нарушений одной из основных функций системы гемостаза. Например, тромбофилия — это патология, при которой повышена склонность к образованию тромбов и внутрисосудистому свертыванию.
Среди первичных естественных антикоагулянтов одним из ключевых является антитромбин III, и дальше речь пойдет о его дефиците и о программе диагностики.
Впервые норвежский ученый Олаф Эгеберг обнаружил сниженную антитромбиновую активность у членов одной семьи с венозным тромбозом в 1965 году, тогда же ее связали с аутосомно-доминантным типом наследования. Позднее идентифицировали и другие виды наследственных тромбофилий. Сейчас с такой патологией живет около 10% общей популяции, а до 40% пациентов, у которых развился тромб, имеют генетические причины.
Недостаточность антитромбина III — наиболее важного ингибитора тромбина — вызывается мутациями гена SERPINC1. Их выявляют у 0,5–2% пациентов с венозной тромбоэмболией.
Дефицит антитромбина III может наследоваться как по аутосомно-доминантному, так и по аутосомно-рецессивному механизму. В общей популяции он встречается в одном случае из 2–5 тыс чел, среди пациентов с тромбозами — в одном из 20–200 случаев. Примерно у половины пациентов развивается один патологический тромб, как правило, после подросткового периода.
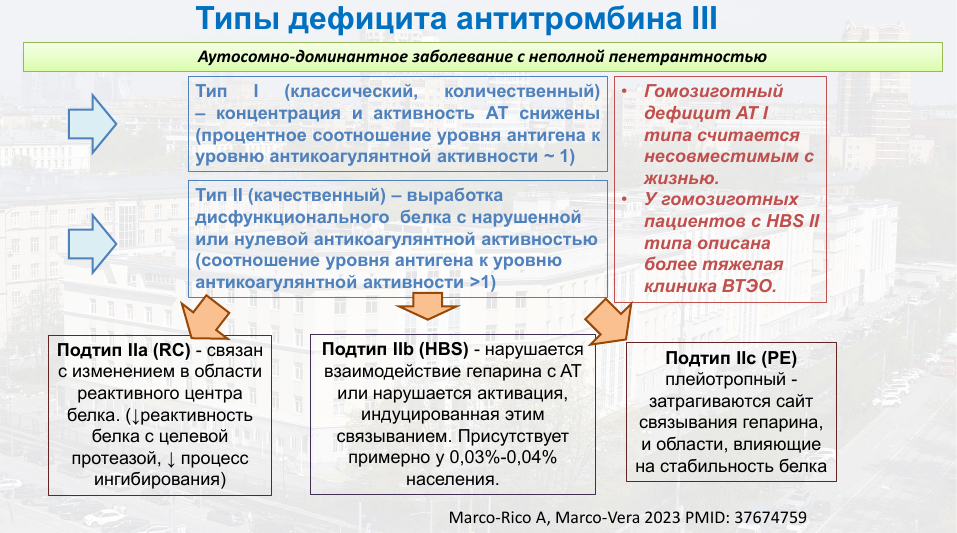
Существует два типа дефицита антитромбина III. Первый из них — количественный, при котором снижена концентрация этого белка и из-за этого снижена антитромбиновая активность. Гомозиготный дефицит антитромбина первого типа считается несовместимым с жизнью.
Второй тип — качественный — связан с выработкой дисфункционального белка и делится на три подтипа. IIa затрагивает реактивный центр, участвующий в ингибировании целевой протеазы, IIb или IIIc нарушают связывание с гепарином или стабильность белка.
До 80% пациентов с дефицитом антитромбина III имеют патогенные каузальные варианты в гене SERPINC1, наследуемые аутосомно-доминантно. Сейчас для этого гена описано более 400 патогенных вариантов. Нонсенс и фреймшифт мутации преимущественно приводят к дефициту антитромбина первого типа, а миссенс-мутации — второго.
Сам ген SERPINC1 состоит из семи экзонов и кодирует белок из 465 аминокислотных остатков; 2-й и 6-й экзоны кодируют основные функциональные домены антитромбина. Из-за довольно простой структуры гена в изначальной программе скрининга мутации в нем выявляли секвенированием по Сэнгеру.
Основным критерием для включения в программу селективного скрининга, запущенную в 2013 году, было подозрение на дефицит антитромбина III и стойкое снижение антитромбиновой активности ниже 70%, также учитывали семейный анамнез. Отбор пациентов в программу проводил лечащий врач, опираясь на данные коагулограммы. Сухие пятна крови пациента направляли в НМИЦ здоровья детей для молекулярно-генетической диагностики.
К настоящему времени на диагностику было направлено 195 пациентов в возрасте от 2 месяцев до 63 лет. Из них у 89 человек выявили каузальные варианты в гене SERPINC1, эти пациенты были из 71 неродственной семьи. Очень важно, что диагностика выявила патологию у 39 детей — при раннем выявлении выше шанс предотвратить развитие патологического тромба.
В целом было выявлено 50 каузальных вариантов в SERPINC1, причем среди них преобладали миссенс-мутации, а 37 этих вариантов (74%) ранее не были описаны, что делает исследование значимым с научной точки зрения и пополняет базы данных.
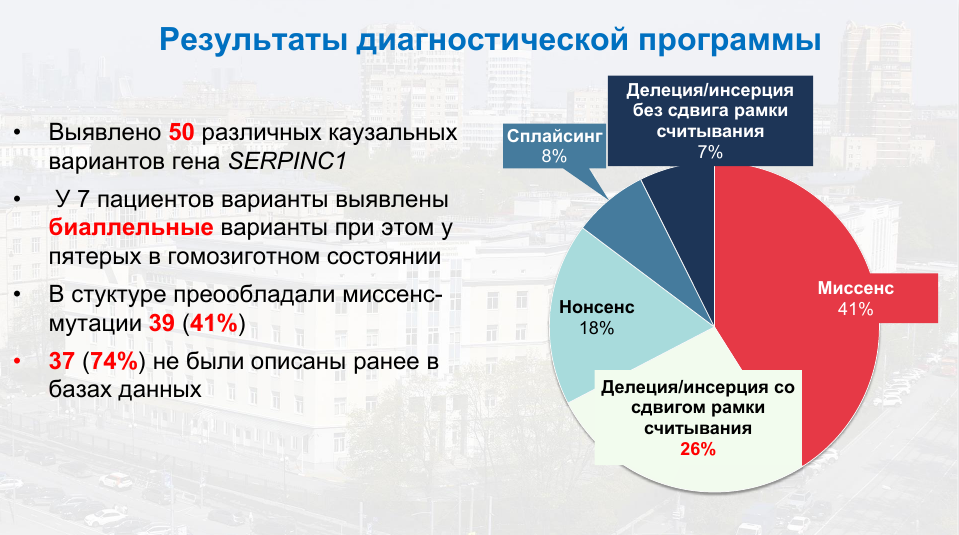
Большинство выявленных вариантов было классифицировано как приводящие к первому типу дефицита антитромбина III — 31 мутация, еще 9 классифицировались как причины второго типа, а для классификации оставшихся требуются дополнительные функциональные исследования.
Пилотно с этого года начали проводить диагностику методами высокопроизводительного секвенирования, и оказалось, что среди 50 пациентов, в том числе исследованных ретроспективно, у троих нашли протяженные делеции или дупликации, хотя ранее причинные варианты у них не выявлялись. Это позволяет исключить проведение MLPA как дополнительный этап додиагностирования, то есть оптимизировать алгоритм и устанавливать лабораторный диагноз в 95-100% случаев.
Ольга Щагина, д.м.н., доцент ФГБНУ МГНЦ им. Н.П. Бочкова, рассказала о случаях, когда фенотип пациента обусловлен комбинацией разных наследственных моногенных заболеваний.
Приход NGS в нашу жизнь принес нам много всего интересного, в том числе и с точки зрения диагностики. На сегодняшний день мы наблюдаем размытие понятия моногенной болезни — модель, согласно которой одна мутация объясняет фенотип, если не устаревает, то по крайней мере уже не очень соответствует действительности. Двойные находки обнаруживаются в 1-5% всех случаев диагностики при экзомном секвенировании и примерно в 1,8% — при диагностике геномным секвенированием.
У 3-4% пациентов, направленных на экзомное или геномное секвенирование, встречаются случайные находки, связанные с генами из списка ACMG. Это самый простой случай, так как речь идет о заболеваниях, для которых существует терапия. В отношении таких находок требуется консультация с участием пациента и членов его семьи, поскольку у них может иметься риск той же патологии.
Следующий вариант — это сочетание двух фенотипов наследственных заболеваний, диагностированных пациенту. Такой вариант встречается примерно у 2% пациентов, и генетические причины болезней могут быть выявлены по отдельности или в рамках одного исследования. Риски рассчитываются для каждого заболевания независимо, а назначать лечение необходимо с учетом всех механизмов патогенеза — в том числе учитывая, что подходы к лечению разных болезней могут взаимодействовать между собой.
Фенотип заболевания также может быть обусловлен сочетанием вариантов в разных генах. Примером служит комбинация двух нервно-мышечных заболеваний — у пациента, как и у отца, имелись нарушения походки, а анализ выявил повышенную активность креатинкиназы, что наводит на мысль о мышечной дистрофии. При этом на электронейромиографии обнаружились признаки демиелинизирующей полинейропатии, и дальнейший молекулярно-генетический анализ подтвердил оба этих диагноза.
В данной ситуации также следует четко разграничивать, действительно ли причиной болезни являются выявленные варианты в разных генах, и обязательно подтверждать их патогенность.
Еще один непростой случай связан с семейными наследственными болезнями, при которых фенотипы, обусловленные вариантами в разных генах, могут комбинироваться при передаче этих вариантов от родителей к детям.

Девочка — гетерозиготная носительница варианта в гене CYBB, связанного с иммунодефицитом, имела легкие проявления заболевания. Семья обратилась за диагностикой носительства X-сцепленного иммунодефицита, когда сестры пробанда планировали беременность. Однако на приеме у сестер обнаружились аномалии кистей и стоп, такие же аномалии были выявлены у их отца. При полногеномном секвенировании оказалось, что у отца и у одной из сестер причиной этих аномалий являются гетерозиготные варианты в гене ROR2, а у еще одной сестры — мутация в гене TP63. При этом носительства иммунодефицита, за диагностикой которого семья обратилась изначально, больше ни у кого не было выявлено.
Из-за того, что не во всех случаях отсутствие сегрегации вариантов в семье с заболеванием свидетельствует об отсутствии причинности этих вариантов, а иногда в одной и той же семье могут встречаться совершенно разные генетические причины одной болезни. Поэтому необходимо аккуратно и внимательно рассматривать фенотип каждого пациента, и все случаи, не укладывающиеся в классический фенотип, должны быть подробно охарактеризованы клинически до назначения молекулярной диагностики. Девиз «мы лечим не болезнь, а больного» справедлив и здесь — мы должны рассматривать именно случай конкретного пациента.
И, конечно же, то, чего мы дождались — внедрение в медицинскую генетику технологий NGS — расширило понимание молекулярных основ фенотипа для каждого пациента и принесло много знаний о пенетрантности и экспрессивности различных генетических вариантов.
Владимир Назаров, к.м.н., Научно-Исследовательский Центр молекулярной медицины, рассказал о молекулярно-генетической диагностике экспансионных болезней.
Внедрение полногеномного и полноэкзомного секвенирования дало нам инструмент для выявления так называемых малых патогенных вариантов. Однако существенную часть диагнозов занимают экспансионные заболевания.
Около 14% генома человека занимают нуклеотидные повторы, увеличение их количества выше референсного значения может приводить к патологиям — это увеличение и называют экспансией.
На сегодня описано около 50 болезней экспансий, которые принято классифицировать по типу наследования, локализации и мотивам повторов, по тому, как эти повторы влияют на кодирующую часть.
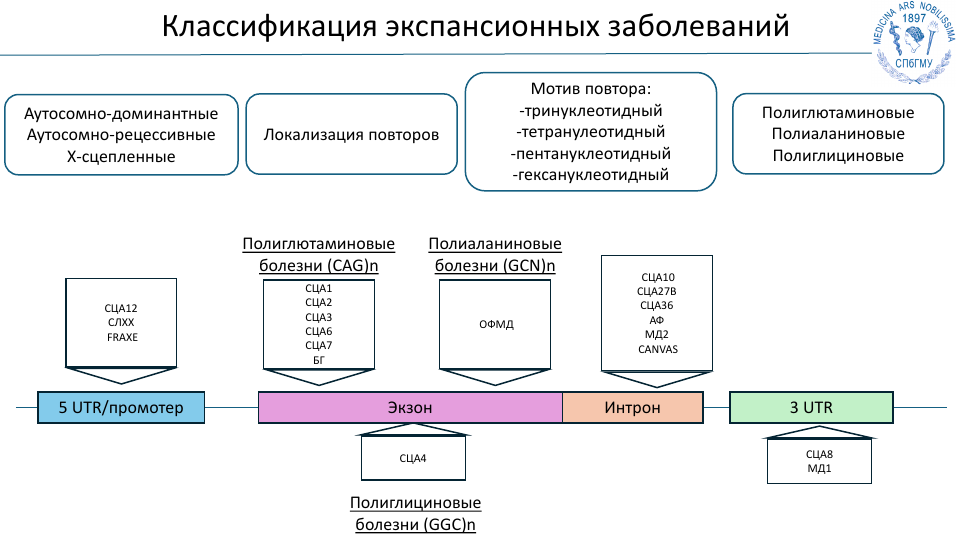
Классические методы NGS не всегда подходят для диагностики болезней экспансии, и в рутинной практике сегодня применяется фрагментный анализ — ПЦР с фланкирующими праймерами или с праймингом тройных повторов. Также могут применяться методы секвенирования третьего поколения, например, нанопоровое.
Особенность молекулярной диагностики, которую важно учитывать — это вариация детектируемого числа повторов, связанная с техническими особенностями и с длиной самого повтора (если число повторов составляет более 42, погрешность возрастает). Влиять на результат могут также особенности синтеза праймеров, например, выбор спейсера для присоединения к праймеру флуоресцентной метки.
При болезнях экспансии встречаются три формы патогенеза — это потеря функции белка, приобретение белком токсических свойств и РНК-токсичность.
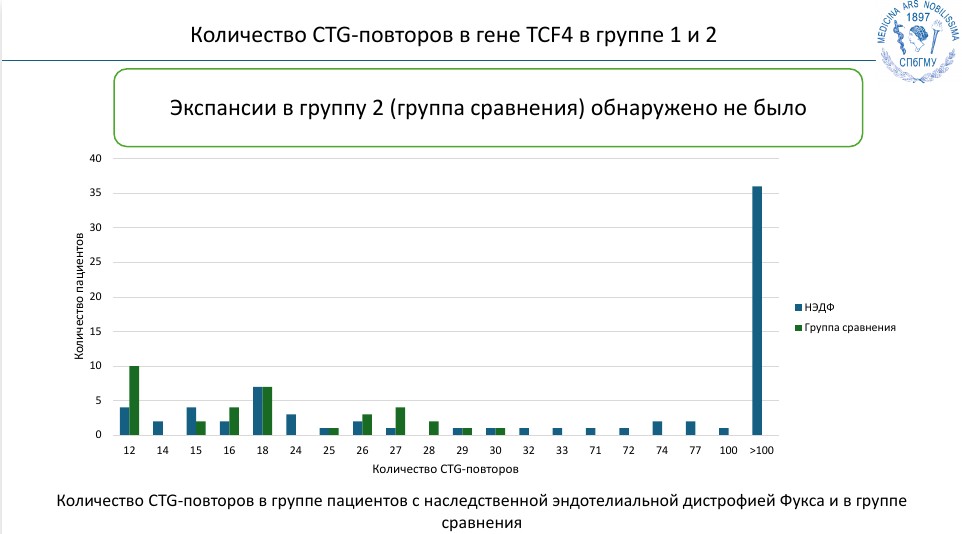
Болезни экспансии кажутся редкими, но на деле их распространенность сильно варьирует. Считается, что в среднем они возникают у 1:3 000 человек, но последние данные указывают на то, что эта цифра занижена почти на порядок. И существует не неврологическое экспансионное заболевание, которое характеризуется крайне высокой распространенностью — это наследственная эндотелиальная дистрофия роговицы Фукса (НЭДФ).
НЭДФ характеризуется дегенерацией роговицы и прогрессирующей потерей зрения. Она поражает 4-5% населения старше 50 лет. Генетическая причина большинства случаев оставалась неизвестной, однако в 2012 году вышла статья, сообщавшая о том, что у большинства пациентов имеются экспансии в гене TCF4, напрямую вовлеченные в патогенез.
Чтобы оценить распространенность этого явления в российской популяции, сотрудники НИЦ молекулярной медицины проанализировали группу пациентов, которым по данным биомикроскопии глаза был поставлен диагноз НЭДФ. В контрольную группу включили пациентов с катарактой, которая отличается по патогенезу. И действительно, в группе с НЭДФ у 60% больных выявили экспансию повторов в гене TCF4, но их не обнаружилось ни у одного участника с катарактой. Эти результаты сопоставимы с зарубежными данными, согласно которым экспансии TCF4 встречаются примерно у 70% людей с НЭДФ. Этот клинический пример подчеркивает, что экспансионных заболеваний, скорее всего, намного больше, чем считается.
Отличительная особенность болезней экспансии — корреляция количества повторов с тяжестью симптоматики, хотя она характерна и не для всех таких заболеваний. Примером служит группа спиноцеребеллярных атаксий. Для болезней этой группы характерны различные специфические проявления, зависящие от подтипа СЦА, и только по клинической картине подтип установить практически невозможно.
За довольно долгий промежуток времени (с января 2017 года) в НИЦ молекулярной медицины проанализировали 1272 пациентов с симптомами СЦА, которых направили на исследование для установления точного диагноза. Патологическая экспансия подтвердилась у 101 человека (8% случаев), при этом более половины подтвержденных случаев приходилось на 1-й тип СЦА. Также среди пациентов подтвердили два случая СЦА 36 типа — оказывается, она все же существует в РФ!
При СЦА 1 и 2 типа также удалось показать обратную корреляцию между количеством повторов и возрастом обращения пациента в лабораторию. При этом возраст обращения связан с возрастом манифестации болезни и отражает ее агрессивность.
Помимо корреляции числа повторов с тяжестью симптомов, существует также феномен предэкспансии, при котором количество повторов определяет фенотип заболевания. Примером служит синдром Мартина-Белла, который связан с экспансией CGG-повторов в промоторе гена FMR1. Если количество повторов превышает 200, развивается классическая форма, для которой характерны психомоторные симптомы расстройств аутистического спектра. Если же повторов больше 55, но меньше 200, клиническая картина будет иная — это первичная яичниковая недостаточность у женщин и синдром тремора/атаксии у мужчин.
Еще одно явление — это феномен антиципации, то есть увеличение числа повторов из поколения в поколение одной родословной. Оно может приводить к утяжелению симптомов.
Таким образом, экспансионные болезни — широкая группа заболеваний со своей спецификой, и, по-видимому, число описанных случаев будет расти по мере внедрения в рутинную практику методов секвенирования с длинными прочтениями для молекулярно-генетической диагностики.
Подробнее о наследственных атаксиях, затронутых в предыдущем докладе, и их диагностике с применением полногеномного секвенирования рассказал Тимофей Визеров, врач-лабораторный генетик из ФГБНУ МГНЦ им. Н.П. Бочкова.
Атаксии — довольно своеобразная совокупность симптомов, которые являются следствием поражения нервной системы на различных уровнях. Определение этого уровня у конкретного пациента важно, поскольку позволяет направить дальнейший диагностический поиск.
Атаксии подразделяются на первичные (к ним относятся наследственные формы) и вторичные, вызванные широким спектром причин и более распространенные. Перед направлением на генетическое обследование важно исключить все возможные причины вторичных атаксий.
Наследственные атаксии — гетерогенная группа заболеваний, и основной интерес для генетической диагностики представляет поиск причинных вариантов. Одну группу причин составляют экспансии повторов, для их диагностики используются таргетные методы. Также причиной болезни могут быть одиночные варианты или геномные перестройки, для диагностики которых предпочтительным методом является массовое параллельное секвенирование. Из-за разницы диагностических стратегий в таких случаях очень важна полнота клинической картины.
Секвенирование генома всегда оставалось методом последней надежды для тех пациентов, у которых другие методы диагностики не выявляли причину болезни, но сейчас благодаря сотрудничеству с «Биотехнологическим кампусом» в рамках проекта «100 000+Я» появилась возможность использовать полногеномное секвенирование как диагностику первой линии.
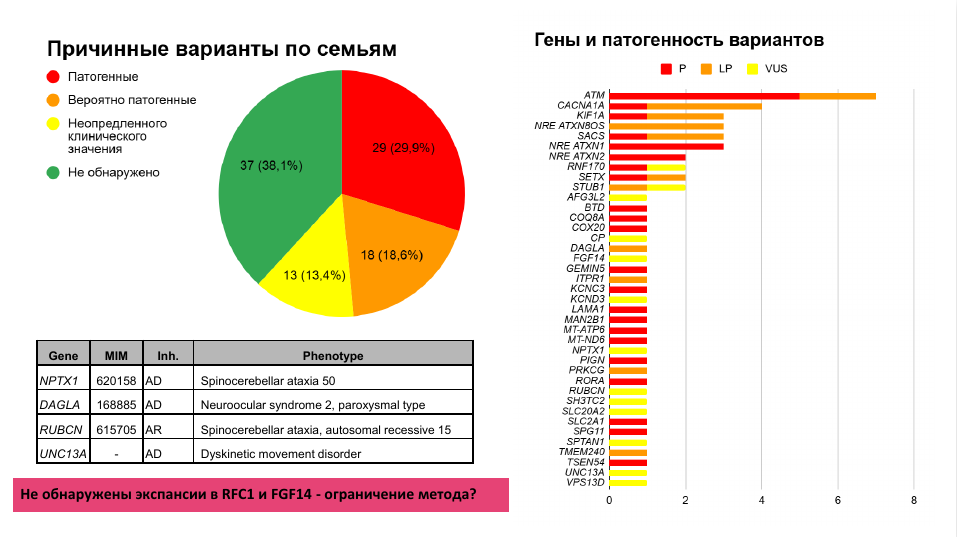
За последние два года в МГНЦ им. Н.П. Бочкова накопились данные, которые подтверждают эффективность полногеномного секвенирования для диагностики наследственных атаксий. В выборку пациентов, которых направляли на генетическую диагностику с подозрением на наследственную первичную атаксию, вошло 197 человек (из них 106 симптомных пациентов) из 97 семей. Часть исследований проводилась в формате «трио». Патогенные и вероятно патогенные варианты были выявлены почти в половине семей (29,9% и 18,6% соответственно), в 38% случаев генетическая причина не была выявлена, однако остается надежда, что у части таких пациентов ее удастся установить с помощью секвенирования длинных прочтений, которым пока проанализировали только 19 человек из выборки.
Отдельно можно выделить спиноцеребеллярную атаксию 8 типа (СЦА8) — аутосомно-доминантное заболевание с выраженной неполной пенетрантностью, вызванное экспансией CTG-повторов. Считается, что в 82% случаев экспансия повторов наследуется от члена семьи без симптомов.
Собственный анализ 17 тысяч пациентов, обращавшихся по другим причинам, подтвердил, что у многих носителей экспансии повторов отсутствуют симптомы. Их распространенность среди здоровых людей составила 0,32%. Патогенная длина повторов пока остается под вопросом, кроме того, существуют разные модификаторы пенетрантности, например, прерывания в области повторов.
Таким образом, полногеномное секвенирование как первая линия — высокоэффективный метод для поиска причин наследственных атаксий, но для валидации подхода необходимо расширять выборку пациентов и накапливать больше данных. Для этого требуется объединение усилий специалистов по всей стране.
Взгляд с другой стороны на наследование моногенных заболеваний с другой стороны предложила аудитории Анна Степанова, к.м.н., ФГБНУ МГНЦ им. Н.П. Бочкова. Она выступила с докладом об однородительской дисомии как причине гомозиготности при рецессивных болезнях.
Однородительская дисомия (ОРД) — это состояние, при котором оба гомолога хромосомной области наследуются только от одного родителя. Ее степень может варьировать от небольшого сегмента до полной хромосомы. ОРД бывают материнского и отцовского происхождения, также разделяют варианты изодисомии и гетеродисомии — в первом случае от одного родителя ребенку передается удвоенная копия одной хромосомы, в другом пара хромосом.
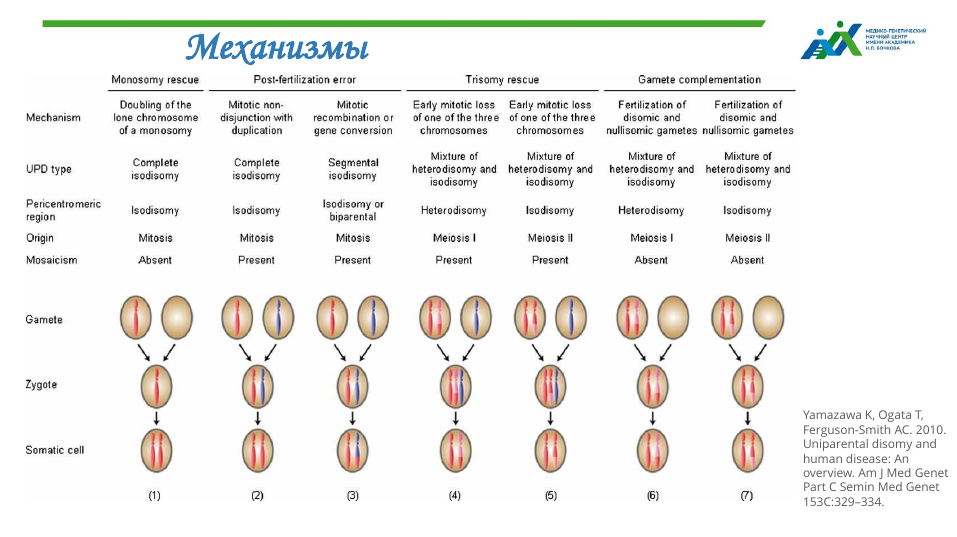
В 1988 году был описан первый случай аутосомно-рецессивного заболевания в результате ОРД — ребенок с муковисцидозом унаследовал обе хромосомы с патогенным вариантом от матери-носительницы.
В 2019 году была определена частота ОРД целых хромосом — примерно 1:2000 среди новорожденных в выборке здоровых. Среди больных с подозрением на генетическую патологию она составила 1:500, при нейродегенеративных болезнях — 1:350.
ОРД чаще всего рассматривались в свете геномного импринтинга, однако появляется все больше исследований, посвященных ОРД как причине аутосомно-рецессивных заболеваний. В МГНЦ было выявлено несколько таких случаев.
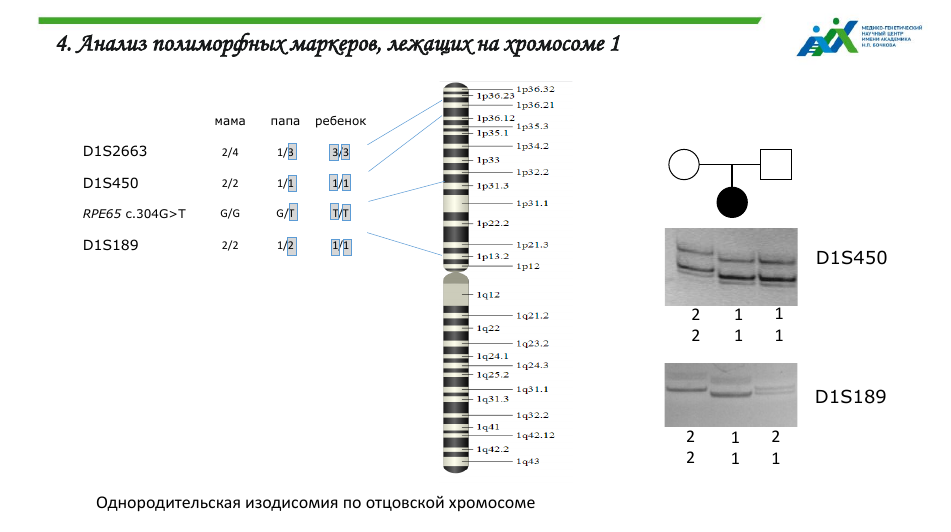
У больного с амаврозом Лебера при секвенировании был выявлен патогенный вариант в гене RPE65. При этом у матери такого варианта не обнаружили, и первым предположением стало то, что мать — носительница крупной делеции в этом участке, поэтому у ребенка патогенный вариант не в гомо-, а в гемизиготном состоянии. Однако анализ числа копий гена RPE65 опроверг это предположение.
На следующем этапе было подтверждено родство ребенка с обоими родителями, и после этого было выдвинуто предположение об однородительской дисомии. Анализ полиморфных маркеров, лежащих недалеко от гена RPE65, показал, что у больного ребенка отсутствуют аллели материнского происхождения. Это подтверждает предположение об ОРД.
Позднее было выявлено еще пять похожих случаев — амавроз Лебера у двух пациентов и по одному случаю врожденной миастении, пигментной дегенерации сетчатки и поясно-конечностной мышечной дистрофии.
Установление истинного механизма наследования особенно важно для медико-генетического консультирования, поскольку важно понимать, что ОРД любой хромосомы может привести к развитию заболевания. Носитель любой рецессивной мутации, связанной с патологией, может стать причиной заболевания у своего ребенка в результате ОРД.
Заключительный доклад в секции прочитал доктор Миньсянь Ван (Minxian Wang), профессор Китайского национального центра биоинформации. Его выступление было посвящено сердечно-сосудистым заболеваниям, которые становятся причиной около половины всех смертей ежегодно. Среди наиболее распространенных — ишемическая болезнь сердца и фибрилляция предсердий.
Одна из важных стратегий предотвращения заболеваний — это анализ семейной истории и наследуемости, для которого в том числе применяется генетическая диагностика. Стоимость секвенирования сейчас быстро снижается, опережая закон Мура. Однако вступая в геномную эру и получая необходимую генетическую информацию, мы также должны понимать, какой вклад в развитие заболевания вносит генетика, а какой — среда.
Среди факторов риска сердечно-сосудистых заболеваний выделяют как генетические, так и средовые. По данным биобанка Японии (Japan Biobank), наследуемость ишемической болезни сердца составляет 50-60%, наследуемость фибрилляции предсердий — около 60-62% по данным UK Biobank.
В контексте сердечно-сосудистых заболеваний можно рассматривать несколько вариантов их генетической архитектуры. Простейшая модель — моногенное наследование — объясняет лишь малую часть случаев. Это связано с тем, что моногенные болезни, как правило, связаны с мутациями в кодирующих участках — то есть в малой части генома человека, — и эти мутации сильно нарушают функцию генов. Модель, которая также учитывает неполную пенетрантность и опирается на 81 клинически релевантных гена, также объясняет недостаточную долю случаев.
На самом деле генетическая архитектура ишемической болезни сердца очень сложна. Количество и степень вклада аллелей риска, которые повышают вероятность развития заболевания, сильно варьирует.
Стратификация риска, основанная на носительстве отдельных мутаций моногенной модели, недостаточно надежна, и более точно объяснить генетику ишемической болезни сердца можно с помощью полигенных индексов риска. Около 98% генома человека — некодирующие области — связаны в первую очередь с генетической предрасположенностью к заболеванию, но не гарантируют его развитие. В связи с этим доктор Ван с коллегами разработали полигенную оценку риска ишемической болезни сердца, опираясь на данные полногеномного поиска ассоциаций (GWAS). Статья с результатами опубликована в Nature Medicine.
Для повышения предсказательной способности модели в оценку риска включили данные GWAS когорт с разным этническим происхождением, а также данные о коморбидных болезнях.
Дальнейшая персонализация индекса основана на объединении отдельных генетических факторов риска и «фоновой» генетической предрасположенности, то есть сочетает моногенную и полигенную модели. Это позволило еще повысить точность оценки риска — предварительно модель протестировали на данных пациентов с раком молочной железы, а затем уже на сердечно-сосудистых заболеваниях.
Наконец, версию 4.0 персонализированной оценки риска сердечно-сосудистых заболеваний дополнили информацией о соматических мутациях. Оказалось, что кумулятивный риск, основанный на отдельных генах в сочетании с полигенным индексом риска и соматическими мутациями, способен выявить в 5–10 раз больше людей, потенциально подверженных риску болезни, чем анализ мутаций в кодирующей части. Это дает почву для своевременного вмешательства — например, изменения образа жизни на более здоровый, уменьшающий влияние внешних факторов риска развития сердечно-сосудистых заболеваний.
Информация о докладчиках
Пушков Александр Алексеевич, к.б.н., ФГАУ «НМИЦ здоровья детей» Минздрава России, г. Москва
Щагина Ольга Анатольевна, д.м.н., доцент ФГБНУ МГНЦ им. Н.П. Бочкова, г. Москва
Назаров Владимир Дмитриевич, к.м.н., Научно-Исследовательский Центр молекулярной медицины МЗ РФ, г. Санкт-Петербург
Визеров Тимофей Викторович, ФГБНУ МГНЦ им. Н.П. Бочкова, г. Москва
Степанова Анна Александровна, к.м.н., ФГБНУ МГНЦ им. Н.П. Бочкова, г. Москва
Minxian Wang, Dr, China National Center for Bioinformation; Beijing Institute of Genomics, Chinese Academy of Sciences, China

Вам будет интересно


 2447
2447
 0
0