Меню
Меню





 Все темы
Все темы
МД-2025: Молекулярно-генетические исследования в неврологии. Часть 1
TDP-43 — потенциальный биомаркер неклассических форм бокового амиотрофического склероза, амброксол — известный муколитик — может помочь при неврологических патологиях (однако для клинического применения его требуется оптимизировать структурно или дополнять другими препаратами), а оптимальный подход к молекулярной диагностике наследственных атаксий должен выявлять как длину экспансии, вызвавшей заболевание, так и ее точную структуру. Об этом и многом другом — на первой половине секции «Молекулярно-генетические исследования в неврологии» конференции «Молекулярная диагностика 2025».

Секцию, посвященную молекулярной генетике в неврологии, открыл доклад о биомаркерах бокового амиотрофического склероза (БАС). Его прочитал Денис Шевчук, м.н.с. ФГБНУ «Российский центр неврологии и нейронаук».
БАС — фатальное и в настоящее время неизлечимое нейродегенеративное заболевание. нейродегенерация. Это самая частая форма болезней двигательного нейрона (около 80% всех случаев). Однако из-за недостатка чувствительных и специфичных маркеров постановка диагноза порой затягивается на год.

Сотрудники РЦНН провели исследование, целью которого было выявить и оценить молекулярно-генетические маркеры БАС. Для этого отобрали 100 пациентов с уже диагностированным БАС и 20 контрольных участников без неврологических заболеваний. Всем им провели мультиплексный анализ на биомаркеры в крови и спинномозговой жидкости (СМЖ), секвенировали ген SOD1, анализировали изоформы TDP43, а также генотипировали экспансии в гене c9orf72.
Пациентов разделили на подгруппы по времени дебюта (ранний, средний и поздний возраст появления первых симптомов), клинической форме (степени вовлечения верхнего и нижнего мотонейрона) и темпу прогрессирования заболевания.
Молекулярно-генетическое тестирование показало, что восемь человек из выборки оказались носителями каузальных мутаций в генах SOD1 (четыре случая), c9orf72 (три случая) или FUS. Число повторов в c9orf72 во всех случаях превышало порог патогенности для БАС.
По профилю биомаркеров пациенты с БАС значимо отличались от контрольных участников. В частности, ROC-анализ показал пороговые значения кластерина до 100% специфичности разделения здоровых контролей и пациентов с БАС.
Концентрация ApoE4 и нейрогранина отличала пациентов с преимущественным вовлечением верхнего мотонейрона (ВМН) от классического фенотипа БАС. Данные изменения могут отражать более выраженное кортикальное повреждение при преимущественном вовлечении ВМН.
Уровни FGF21 были связаны с формой БАС — этого биомаркера было больше при бульбарной форме, чем при шейно-грудной или крестцовой. Однако такую анатомическую зависимость объяснить сложно. Возможно, это связано с более высоким вкладом нейровоспаления при более агрессивной форме.
Наконец, концентрации кластерина и факторов комплемента H (CFH) и C3 отличались у пациентов с разным темпом прогрессии БАС. Уровень CFH был выше при быстрой прогрессии заболевания, и это впервые показали на достаточно большой выборке. Полученный результат заставляет задаться вопросом, не может ли вмешательство в систему комплемента стать способом модуляции процессов патологии.
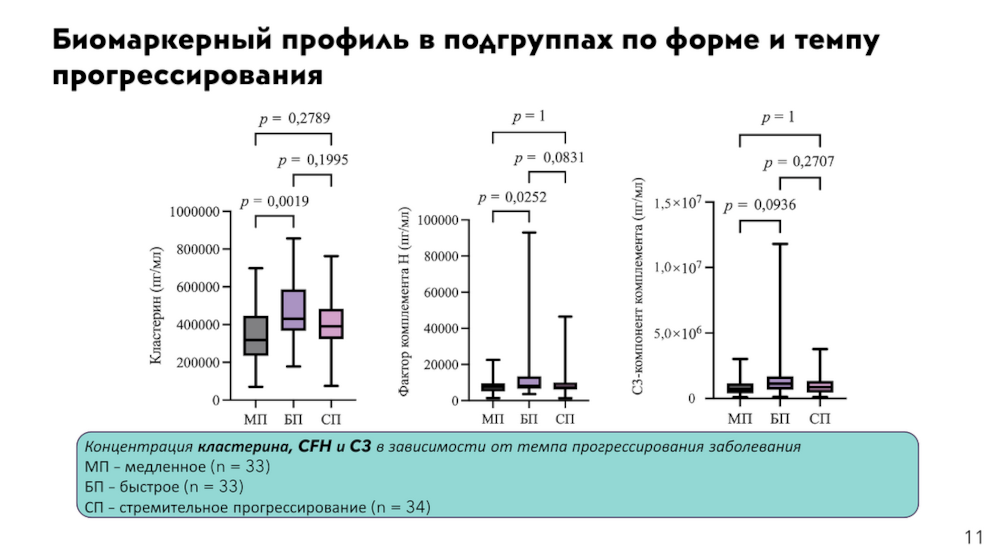
По результатам ROC-анализа кластерин сочли наиболее перспективным биомаркером. Предположительно, этот шаперон непосредственно участвует в патологии и гибели мотонейронов.
TDP-43 является одной из главных детерминант патофизиологии при БАС. TDP-43 протеинопатии выявляются у большинства пациентов со спорадической формой и даже при некоторых наследственных, и в 97–98% они развиваются даже без мутации в его гене. Протеинопатия при БАС включает сразу два механизма — нарушение ядерной локализации TDP-43 и приобретение им нежелательных функций в цитоплазме.
TDP-43 склонен к прионоподобной конверсии, которая возможна за счет С-концевого домена низкой сложности. Его патологические формы могут распространяться по нервной системе различными путями межклеточной коммуникации, приводя к распространению нейродегенерации.
Для детекции патологических изоформ TDP-43 с прионоподобной активностью воспользовались методом затравочной амплификации. Такой анализ спинномозговой жидкости участников исследования в реальном времени выявил патологические изоформы и подтвердил потенциал TDP-43 как биомаркера БАС и затравочной амплификации как метода детекции, в том числе для выявления неклассической формы БАС.
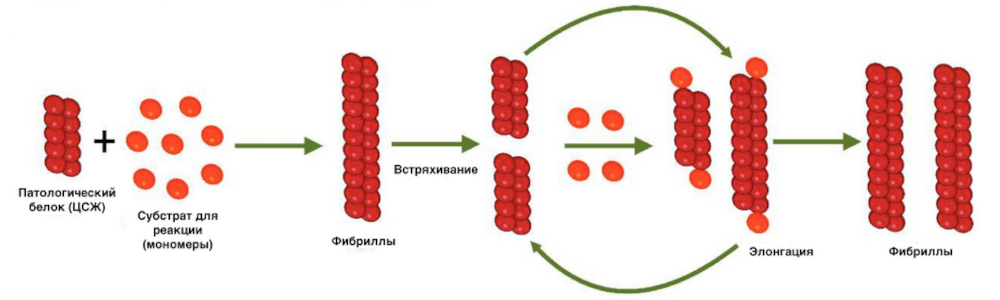 Реакция затравочной амплификации.
Реакция затравочной амплификации.
С докладом о стратегиях детекции экспансии нуклеотидных повторов в гене FGF14 при диагностике наследственной атаксии SCA27B выступила Арина Проценко, ФГБНУ «Российский центр неврологии и нейронаук».
Микросателлиты — повторы 1–6 нуклеотидных мотивов — встречаются на протяжении всего генома человека и занимают около 3%. Их длины обладают пороговым значением — выше определенного числа повторов возникает патология. Это состояние называют экспансией нуклеотидных повторов.
Наиболее часто встречаются при нейродегенерациях экспансии мотива CAG. Еще один из вариантов — повторы GAA, которые, например, характерны для спиноцеребеллярной атаксии (СЦА) типа 27B, которой и посвящен дальнейший анализ.
От числа повторов зависит возраст дебюта и тяжесть течения многих неврологических заболеваний, связанных с экспансиями. Поэтому точное определение их числа — важная, но непростая задача молекулярной диагностики.
В 2023 году было обнаружено, что повторы GAA в гене FGF14 приводят к новому типу СЦА — 27B. Анализ собственной выборки пациентов с врожденными аутосомно-доминантно наследуемыми атаксиями показал, что этот тип составляет около 10% случаев. Пенетрантность СЦА27B зависит от числа повторов — полная начинается при накоплении 300 и более. У некоторых людей встречается повторяющийся мотив GAAGGA вместо GAA, который нивелирует патогенность. Поэтому для корректной диагностики важно не только определять длину экспансии повторов, но и точную структуру.
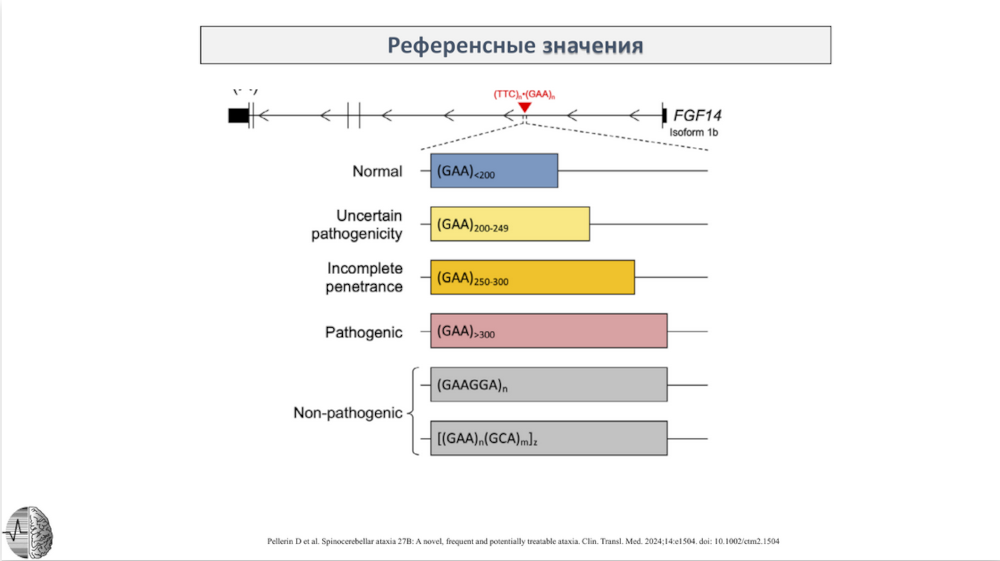
Для решения этой задачи необходим стандартизированный диагностический подход. В рамках пилотного исследования были протестированы следующие стратегии:
-
ПЦР с фланкирующими праймерами и фрагментный анализ. Этот метод позволяет детектировать два непатогенных аллеля и остановить дальнейший скрининг для таких образцов, но имеет ограничения по обнаружимой длине продуктов.
-
PR-PCR. Этот метод позволяет подтвердить структуру нуклеотидных повторов в коротких аллелях, в том числе выявить прерывание повтора, если оно есть. Однако он также имеет ограничения по длине до 120 повторов.
-
Секвенирование по Сэнгеру — подход, с помощью которого можно определить повторяющийся мотив экспансии, но не секвенировать экспансированные аллели полностью. В данном исследовании секвенирование по Сэнгеру использовалось только в качестве дополнительного метода.
-
Электрофорез в геле или на приборе Qsep лишен ограничения по длине анализируемых повторов, поэтому для данной задачи это предпочтительный метод (и Qsep обеспечивает более точное определение). Однако этот подход не отражает их структуру.
С помощью секвенирования по Сэнгеру провели анализ вариативного участка, лежащего в 5'-области перед повтором. Известно, что эта область отличается у людей с экспансией и без, однако на собственных данных сейчас не удалось подтвердить гипотезу о стабилизации структуры за счет вариативной области. Вариант, который было принято ассоциировать со стабилизацией, нашли и при экспансии повторов.
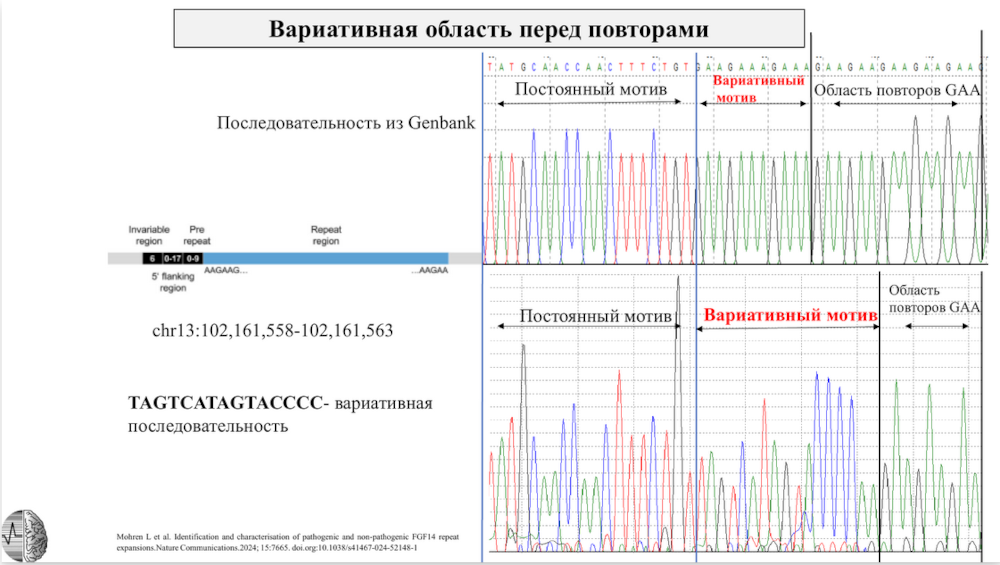
Для корректировки результатов, полученных с помощью Qsep, было решено использовать нанопоровое секвенирование. Полученная комбинация методов и есть итог проведенного исследования — нанопоровое секвенирование признали наиболее предпочтительным вариантом для выявления экспансий повторов при диагностике наследственных атаксий.
Доклад Евгении Мельник (к.м.н., МГНЦ им. Н.П. Бочкова) был посвящен наследственным артрогрипозам при поражении нервно-мышечной системы.
Развитие методов массового параллельного секвенирования дало возможность рассмотреть большие группы пациентов с наследственными патологиями.
Артрогрипоз — это врожденная контрактура двух и более уровней суставов. Существует несколько видов артрогрипозов, отличающихся локализацией поражения — при мышечных страдает сократительная активность мускулатуры, при нервно-мышечных нарушается работа нервно-мышечного синапса; также существуют варианты с поражением сенсорных или моторных нейронов.
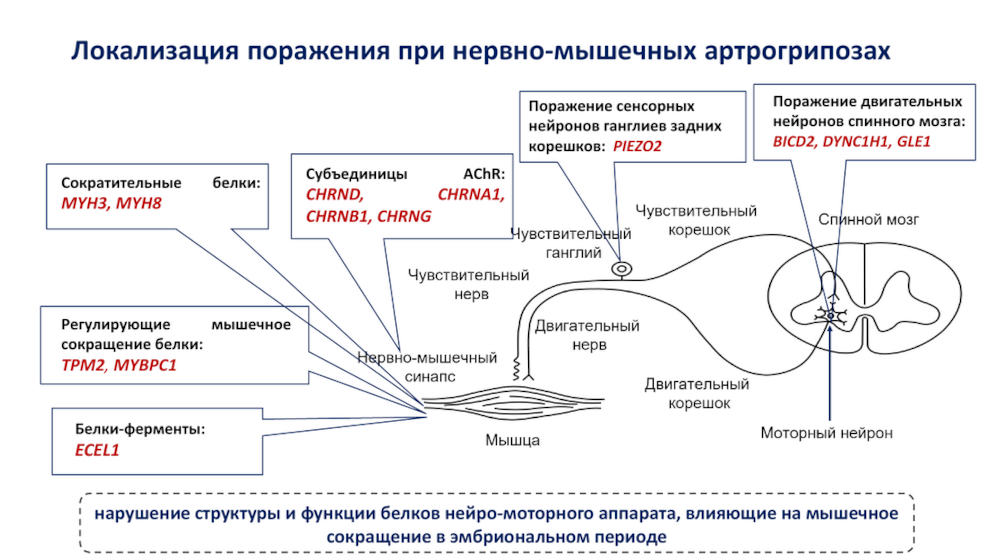
Дистальные артрогрипозы (ДА) включают 12 форм и возникают в основном при нарушении изоформ белков сократительного аппарата в эмбриональном периоде (в постнатальном периоде аналогичное нарушение приведет уже к миопатии).
Например, дистальный артрогрипоз типа 2А, или синдром Фримена-Шелдона — это аутосомно-доминантное наследственное заболевание, связанное с мутацией в гене MYH3. Этот ген кодирует тяжелую цепь миозина и содержит определенные рекуррентные варианты, что дает возможность для диагностики.
ДА типа 7 — синдром тризма нижней челюсти и псевдокамптодактилии — обладает своей характерной заменой в гене MYH8. Ее выявляли в том числе у пациентов, наблюдавшихся в МГНЦ им. Н.П. Бочкова.
При миопатиях пациенту проводят МРТ мышц всего тела, и сотрудники МГНЦ им. Н.П. Бочкова решили проверить, что покажет МРТ у их пациентов. В частности, они проанализировали случаи заболеваний, связанных с дисфункцией тропомиозина. Это врожденные миопатии, по мере прогрессирования которых развиваются контрактуры суставов. У пациентов с заболеваниями этой группы на МРТ не было выявлено признаков фиброза или замещения мышц жировой тканью.
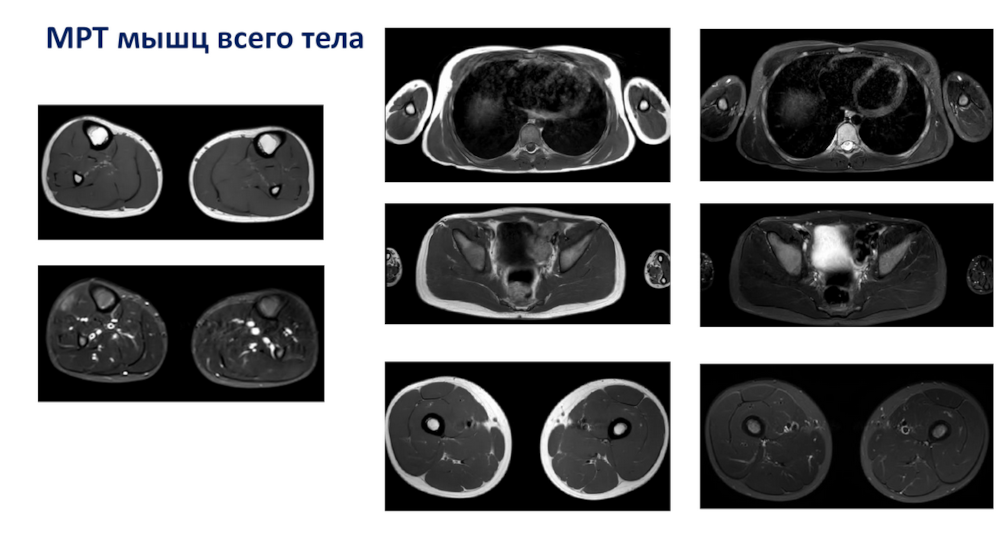
Большинство артрогриппозов данной группы характеризуется аутосомно-доминантным типом наследования, поэтому обычно при подозрении на них оценивают также состояние родителей. Однако есть и случаи, когда, несмотря на наличие доминантного патогенного варианта, проявления симптомов у матери пациента были минимальны и она их не замечала.
Еще один механизм — нарушение активности эндотелин-превращающего белка ECEL1 (фермента мышечного волокна). В этом случае болезнь связана с нарушением формирования нервно-мышечного синапса, а наследование будет рецессивным. Такие пациенты отличаются фенотипически — у них выражена центральная атрофия языка, гиперлордоз, имеются кожные складки. В данной группе МРТ-сканирование уже показало, что мышцы замещаются жиром.
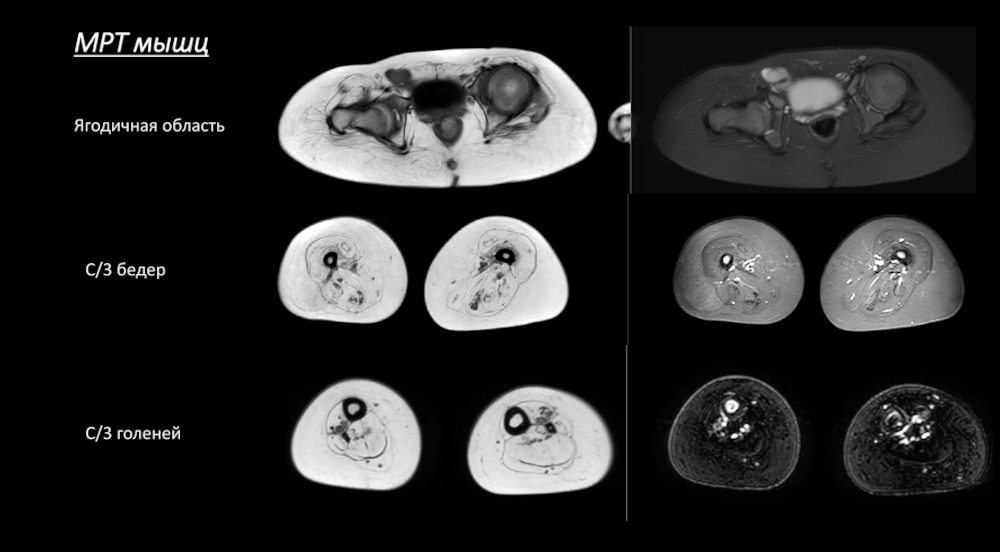
Патология также возможна на уровне ацетилхолиновых рецепторов AChR — замена фетальной субъединицы на взрослую может не происходить.
Наконец, еще одна разновидность — это спинальная мышечная атрофия (СМА) с преимущественным поражением ног (SMALED). Заболевание сопровождается артрогрипозом и врожденными переломами, а его генетическая причина — это мутации в гене DYNC1H1. При SMALED происходит практически полное жировое замещение мышц, такие пациенты наблюдаются у ортопедов и переносят множество оперативных вмешательств.
В общей сложности проведенные на базе МГНЦ им. Н.П. Бочкова исследования выявили наследственные артрогрипозы приблизительно у 60% пациентов из выборки, в которую включили 300 человек.
Таким образом, наследственные артрогрипозы — это генетически гетерогенная и клинически полиморфная группа заболеваний. Из них около 40% обусловлены поражением нервно-мышечного аппарата, при них не происходит структурных поражений мышц.
Из-за небольшого числа часто встречающихся патогенных вариантов при подозрении на ДА типа 2А или типа 7 пациента нужно направлять на секвенирование с целью поиска этих вариантов. Во всех остальных случаях для выявления генетической причины рекомендовано полноэкзомное или полногеномное секвенирование в формате «трио».
Софья Пчелина, д.б.н., НИЦ ПСПбГМУ им акад. И.П. Павлова, г. Санкт-Петербург, рассказала о молекулярной диагностике наследственных форм болезни Паркинсона.
Распространенность болезни Паркинсона (БП) — около 1–2% среди лиц старше 60 лет. Диагноз сейчас ставят неврологи по сочетанию клинических симптомов. И если биомаркеры и нейропротекторная терапия БП пока под вопросом, то ДНК-диагностика наследственных форм этого заболевания уже доступна. Далее речь пойдет о целях этой диагностики и ее алгоритмах.
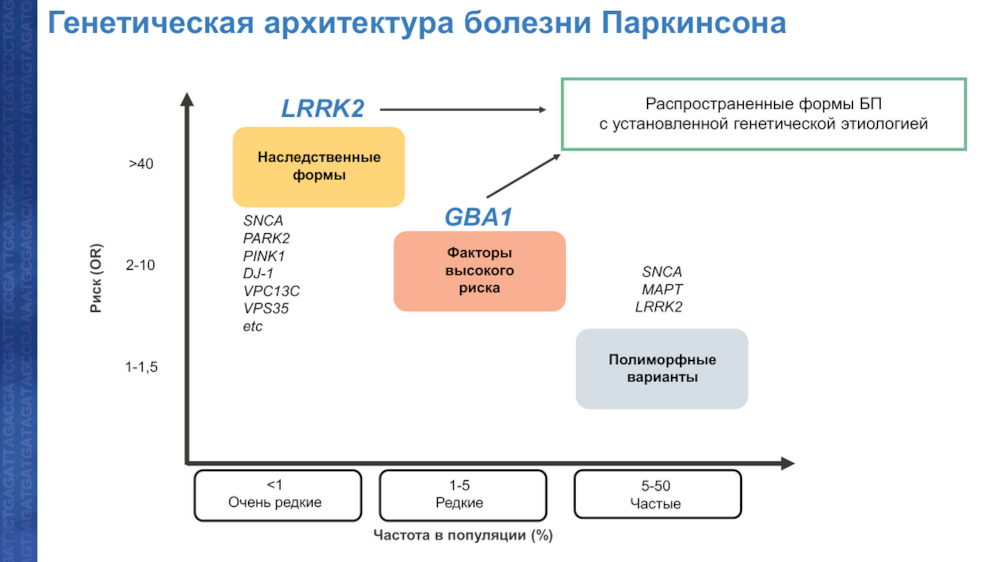
Генетические основы БП достаточно разнородны — существуют моногенные формы, причем как аутосомно-доминантные, так и аутосомно-рецессивные варианты. Также известны многочисленные варианты риска, «однако о них в контексте молекулярной диагностики мы говорить не имеем права», — подчеркнула докладчица. Наконец, встречаются варианты с неполной пенетрантностью.
На базе ПСПбГМУ им акад. И.П. Павлова проводится скрининг мажорных мутаций в LRRK2 и GBA1 методом ПЦР. С помощью такого подхода проанализировали уже собранный биобанк из 2096 пациентов. Тех, у которых мажорные мутации в этих генах выявлены не были, направляли на таргетное секвенирование. Оно охватывало панель из 52 генов, ассоциированных с наследственными формами БП. Наконец, в редких случаях проводилось экзомное секвенирование.
Оценка распространенности наследственных форм БП показала, что наиболее частые случаи связаны именно с мутациями в гене GBA1. Анализ был нацелен на три наиболее распространенных варианта — «тяжелая мутация» (ассоциирована с повышением риска БП до 24 раз, особенно ранних форм), «легкая мутация» (ассоциирована с повышением риска в 8 раз) и «аллель риска» (ассоциирован с повышением риска в 3 раза). Скрининг на мутации GBA1 выявил описанные мажорные варианты у 5,2% человек.
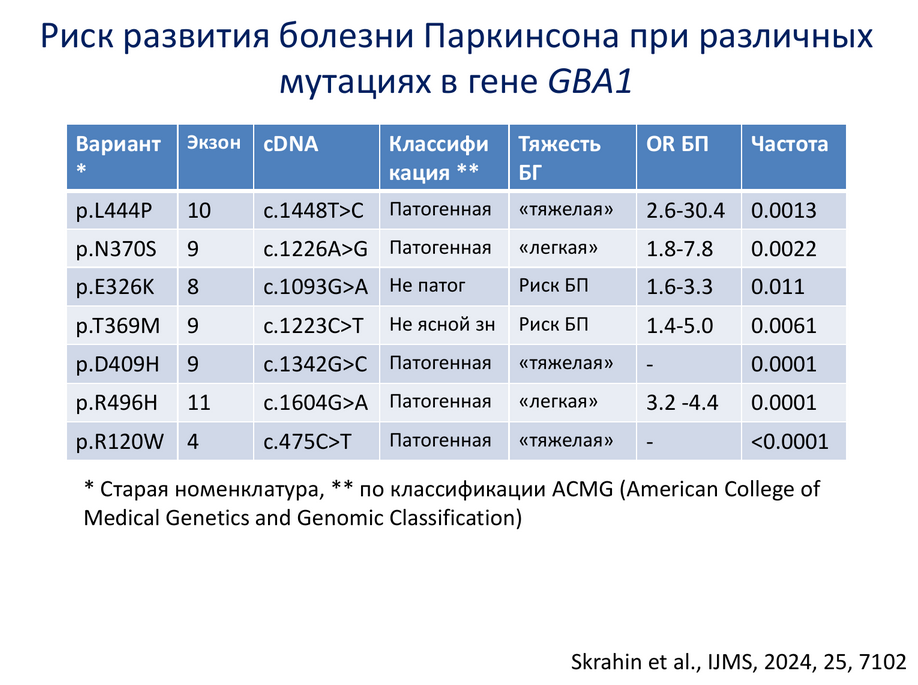
Клиническое течение GBA1-БП отличается от идиопатической формы более ранним началом, высоким риском развития деменции и превалированием неврологических симптомов.
Возникает вопрос, прибавит ли что-то к диагностике наследственных форм БП панель таргетного секвенирования? По опыту данного исследования, в котором на дополнительный анализ направили 497 пациентов, оказалось, что нет — NGS-панель не улучшала выявление генетических причин БП, кроме нескольких отдельных случаев.
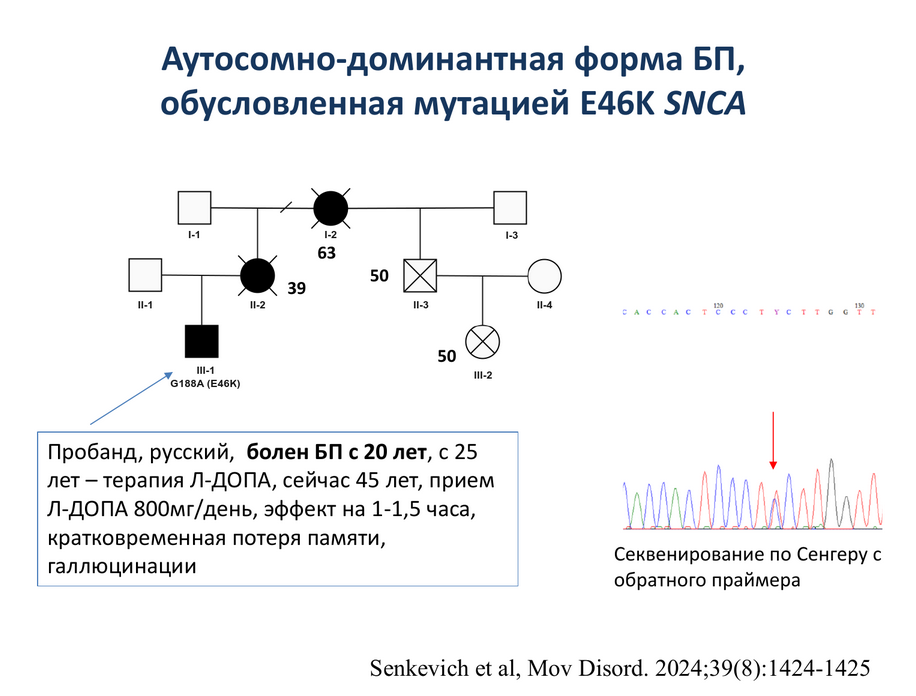
Зато анализ выявил уникальный случай — третий в мире и первый в популяции славянского происхождения. Секвенирование позволило обнаружить семью с альфа-синуклеинопатией, которая вызвана мутацией E46K в гене SNCA. Этот вариант привел к развитию БП у пробанда в возрасте 20 лет.
Как уже говорилось, нейропротекторная терапия на сегодня все еще под вопросом, однако попытки ее разработать продолжаются.
Если причина развития БП — это агрегация альфа-синуклеина, то, вероятно, ее можно ингибировать антителами или малыми молекулами. Препараты антител, которые предотвращали бы агрегацию альфа-синуклеина, провалились в клинических испытаниях, а две малые молекулы сейчас проходят их раннюю стадию.
Другие мишени для терапии — это киназа LRRK2 или глюкоцереброзидаза. Одна из малых молекул — модуляторов активности глюкоцереброзидазы — это амброксол, известный муколитик. Исследование по репозиционированию препаратов выявило его как потенциальный активатор глюкоцереброзидазы, однако он неудобен тем, что при связывании занимает еще и активный сайт фермента. Поэтому ведется поиск аллостерических модуляторов активности и работы по их оптимизации.
Тем не менее, амброксол уже дошел до фазы 3 КИ при болезни Паркинсона и болезни Гоше. Сейчас в России два пациента с БП проходят лечение амброксолом.
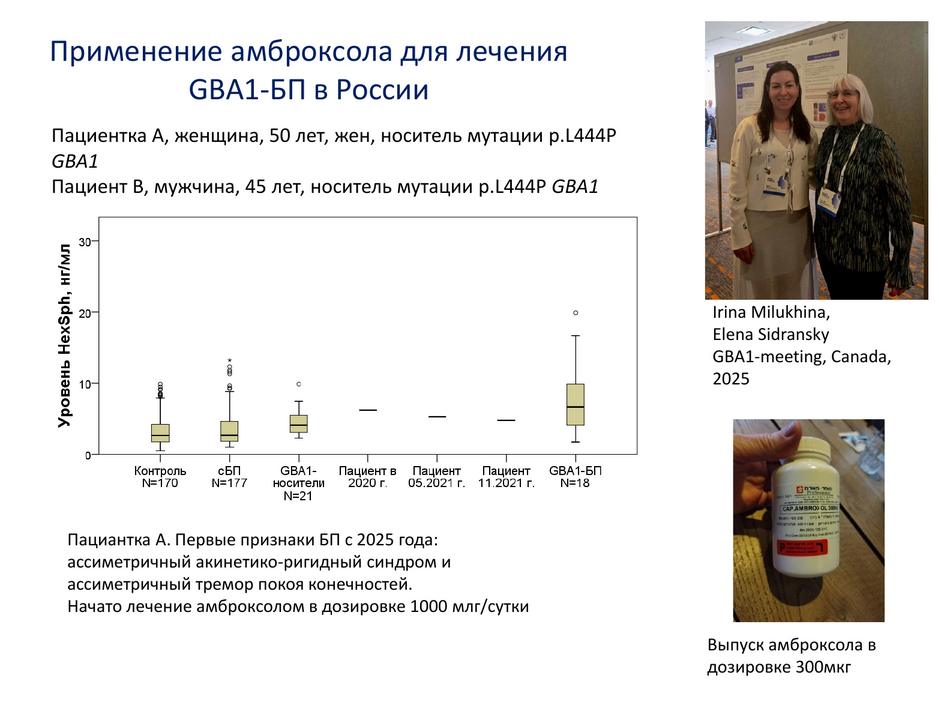
Также ведется разработка аллостерического модулятора активности глюкоцереброзидазы. Выбранные по данным молекулярного докинга структуры тестируют на первичных макрофагах и дофаминергических нейронах, полученных из ИПСК пациентов. Одну такую молекулу Софья Пчелина и коллеги уже запатентовали и в настоящее время проводят оценивание ее нейропротекторных свойств.
Помимо генетического тестирования, можно проводить также поиск биомаркеров в периферической крови. Похоже, что надежных биомаркеров БП в периферической крови не существует, однако некоторые молекулы можно применять их в качестве дополнительного инструмента — например, уровень кислой сфингомилеиназы позволяет отличить ранние стадии БП от мультисистемных атрофий.
Тему лечения БП продолжила Татьяна Усенко, к.б.н., НИЦ ПСПбГМУ им акад. И.П. Павлова. Ее доклад был посвящен разработке таргетной терапии с использованием пациент-специфичных клеток.
У болезни Паркинсона существуют две основные формы, о первой из них — GBA1-ассоциированной — подробно говорилось в предыдущем докладе. Терапия в данном случае направлена на повышение активности фермента, утратившего функцию — глюкоцереброзидазы.
Терапевтический эффект амброксола в данном случае уже подтвердили на дофаминергических нейронах, которые были получены из ИПСК пациентов в рамках сотрудничества с Институтом цитологии и генетики в Новосибирске. Однако в случае с амброксолом есть нюанс — терапия требует высоких доз, кроме того, препарат производится в форме таблеток. Это повышает вероятность возникновения побочных эффектов у пациента, поэтому исследователи задались вопросом, можно ли как-то потенцировать действие амброксола и тем самым снизить эффективную дозировку.
И здесь речь идет о другом механизме развития БП, связанном с патологическим повышением активности киназы LRRK2. Очевидно, что киназа должна фосфорилировать какие-то белки, однако их долго не могли найти. Наконец, методом фосфопротеомного анализа удалось показать, что мишенями в данном случае являются Rab10 и другие Rab-белки, вовлеченные в эндолизосомный транспорт.
Исследователи показали, что ингибирование LRRK2 приводит в том числе к повышению активности глюкоцереброзидазы. Следующим логичным шагом стала проверка того, усилит ли ингибирование LRRK2 действие модуляторов GBA1, в данном случае амброксола. Однако потенцирующего эффекта обнаружено не было.
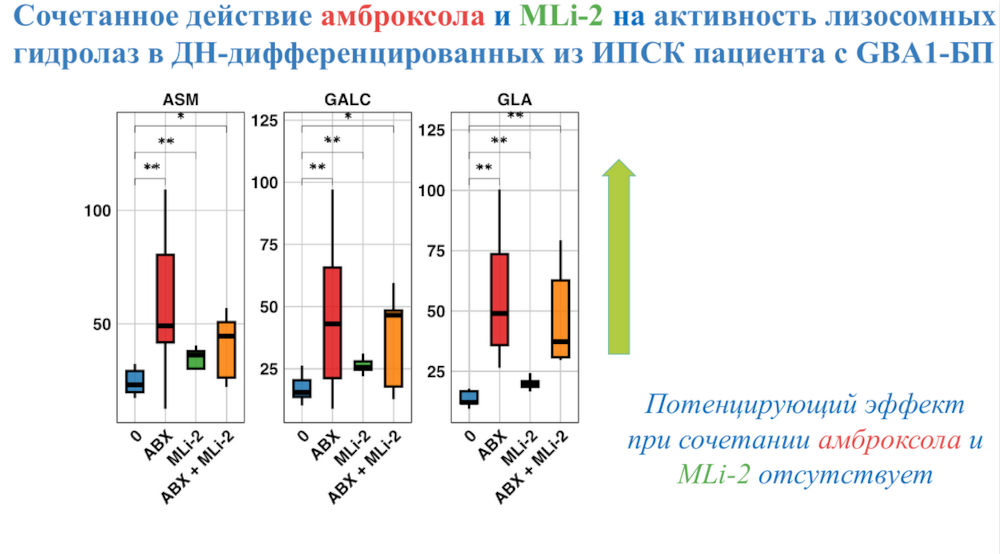
Пытаясь установить причину этого, исследователи показали, что ингибитор LRRK2, как и ожидалось, усиливает эндолизосомный транспорт. Однако амброксол помимо целевой мишени активирует также другие лизосомные ферменты. Оказалось, что фосфорилирование Rab10 растет в присутствии амброксола. Увеличение его фосфорилирования может свидетельствовать о том, что эндолизосомный транспорт замедляется.
На основании дальнейшего анализа исследователи выдвинули предположение, чем может объясняться неэффективность комбинации амброксола с ингибиторами LRRK2. Амброксол способен связываться с мембранами напрямую и за счет этого менять их свойства. В частности, он может менять форму мембраны, которая важна для нормальной работы PPM1H — фосфатазы-антагониста LRRK2. Вероятно, амброксол препятствует функционированию этой фосфатазы, что и ослабляет терапевтический эффект, однако это предположение еще предстоит подтвердить. Тем не менее, полученные данные подчеркивают, насколько важно понимать, нет ли антагонистического действия между компонентами комбинированной терапии.
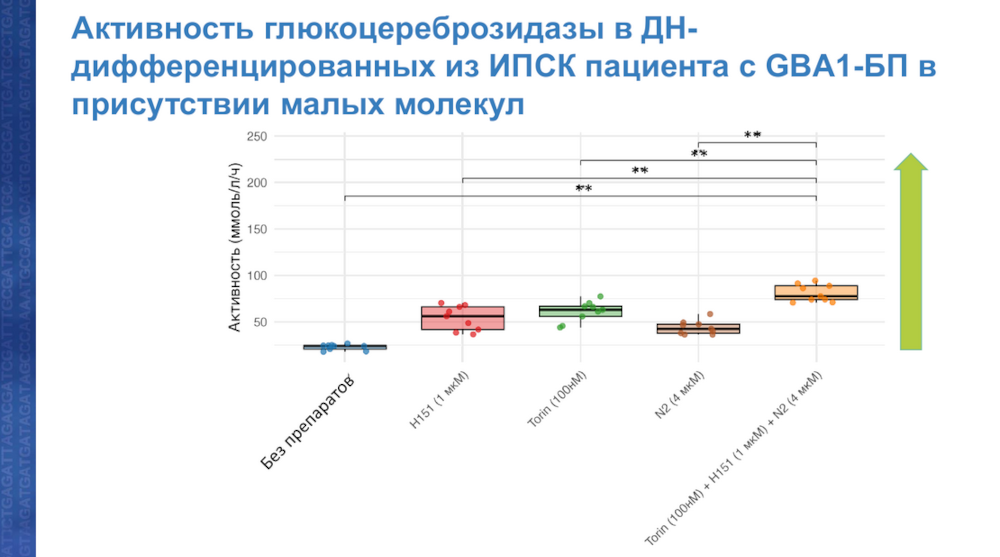
Альтернативный подход основан на вмешательстве в менее специфичные сигнальные пути. Так, при GBA1-БП происходит активация сигнального пути mTOR. Ингибирование этого сигналинга с помощью малых молекул — одно из перспективных направлений разработки терапии БП. Кроме того, при БП усиливается воспаление, которое регулируется белком STING. Уже показано, что ингибирование этого белка соединением H-151 снижает нейровоспаление и нейродегенерацию в мышиной модели.
Сотрудники НИЦ ПСПбГМУ им акад. И.П. Павлова проверили, скажет ли ингибирование STING потенцирующий эффект на терапевтическую активность разработанных ими малых молекул — активаторов глюкоцереброзидазы. На нейронах, полученных из ИПСК пациентов, удалось это подтвердить. Комбинация трех малых молекул, нацеленных на STING, mTOR и глюкоцереброзидазу, действительно усиливала активность последней в модели in vitro.
МД-2025: Молекулярно-генетические исследования в неврологии. Часть 2
Информация о докладчиках
Шевчук Денис Владимирович, к.м.н. ФГБНУ «Российский центр неврологии и нейронаук», г. Москва
Проценко Арина Романовна, ФГБНУ «Российский центр неврологии и нейронаук», г. Москва
Мельник Евгения Александровна, к.м.н., ФГБНУ МГНЦ им. Н.П. Бочкова, г. Москва
Пчелина Софья Николаевна, д.б.н., НИЦ ПСПбГМУ им акад И П Павлова, г. Санкт-Петербург; «Петербургский институт ядерной физики им. Б.П. Константинова», Национальный исследовательский центр Курчатовский институт, г. Гатчина
Усенко Татьяна Сергеевна, к.б.н., НИЦ ПСПбГМУ им акад И П Павлова, г. Санкт-Петербург; «Петербургский институт ядерной физики им. Б.П. Константинова», Национальный исследовательский центр Курчатовский институт, г. Гатчина

Вам будет интересно


 2450
2450
 0
0