Меню
Меню





 Все темы
Все темы
Насколько страшны прионы на самом деле?
Наверное, все так или иначе слышали страшные слова «прионная болезнь». Чем так уникальны прионы, правда ли прионные болезни настолько страшны и неизлечимы и что вообще наука знает о прионах на сегодняшний день? Разбираемся по порядку.
Структура основного прионного белка (PrP).
Credit:
123rf.com
Что вообще такое прион?
Как размножаются эти загадочные инфекционные агенты?
Как можно заболеть (и, главное, как не заболеть) прионными болезнями?
Неужели совсем нет способа выявить опасность, пока не стало совсем поздно?
Как диагностируют болезнь Крейтцфельдта-Якоба у живых людей, без биопсии мозга?
Прионные болезни считаются неизлечимыми. А может, все-таки можно их вылечить?
Устойчивость к прионным инфекциям возможна?
Всегда ли прионы таят в себе опасность?
Если у меня нет мутации, мне можно не бояться?
Слово «прион» ввел в 1982 году доктор медицинских наук Стэнли Прузинер, которому впоследствии присудили Нобелевскую премию за открытие прионов. Оно образовано от слов «белок» (protein) и «инфекция» (infection).
История изучения прионов берет начало в XVIII веке, если отсчитывать ее с описания симптомов первой прионной болезни — почесухи, или скрейпи, у овец.
Немного хронологии:
• 1700-е годы — первые известные случаи скрейпи овец.
• 1920-е годы — описали болезнь Крейтцфельдта—Якоба.
• 1961 год — описали скрейпи у мышей.
• 1960-е — обнаружили, что трансмиссивные губчатые энцефалопатии могут вызываться исключительно белковым агентом.
• 1982 — группа Прузинера впервые выделила и описала прионный белок. Тогда же был введен термин «прион» для обозначения его инфекционной формы.
• 1986 — прионные болезни привлекли внимание широкой общественности из-за эпидемии «коровьего бешенства» (губчатой энцефалопатии крупного рогатого скота) в Великобритании. Как позже выяснилось, она была связана с тем, что прионы из костной муки, которую добавляли в корм крупного рогатого скота, сохранили инфекционные свойства и распространились среди животных.
• начало XXI века — случаи передачи «коровьего бешенства» людям. Сейчас инфекция у человека, вызванная такой формой прионного белка, считается новой формой болезни Крейтцфельдта—Якоба
Что вообще такое прион?
Прионы как инфекционные агенты представляют собой аномально свернутую форму основного прионного белка (PrP) — кодирующий его ген экспрессируется во всех здоровых тканях людей и различных животных (прионные белки также встречаются у грибов и, вероятно, растений, но об этом позже).
PrPC — нормальная форма — это мембранный белок млекопитающих. Его функция изучена недостаточно; по некоторым данным, он участвует в передаче сигналов в клетке, а также способствует миелинизации аксонов в нервной системе.
Инфекционная форма обозначается PrPd или PrPSc (индексы обозначают, соответственно, слово disease — «болезнь» — или Scrapie — название болезни скрейпи овец, которая считается первым описанным прионным заболеванием). Она имеет иную пространственную структуру, за счет которой весьма стабильна и устойчива к протеазам, поэтому практически не подвергается расщеплению в организме. В гене PRNP, кодирующем основной прионный белок человека, описан ряд мутаций, которые ассоциированы с различными прионными болезнями и, по-видимому, способствуют сворачиванию PrP в патогенную форму.
Однако распространение прионов связано не только с мутацией в этом белке.
Как размножаются эти загадочные инфекционные агенты?
По наиболее распространенной гипотезе, молекулы PrPd способны катализировать переход нормального белка в прионную форму. Предложено несколько моделей, объясняющих детали этого механизма, но суть сводится к одному — аномально свернутая молекула (одна или в составе фибриллы) взаимодействует с нормальным белком, который при этом изменяет конформацию и превращается в инфекционную, гораздо более стабильную форму. Как раз из-за ее высокой стабильности процесс однонаправленный — нормальный прионный белок не может катализировать обратный переход PrPd в безопасную неинфекционную форму. Поэтому патогенный вариант белка накапливаются в клетке экспоненциально.
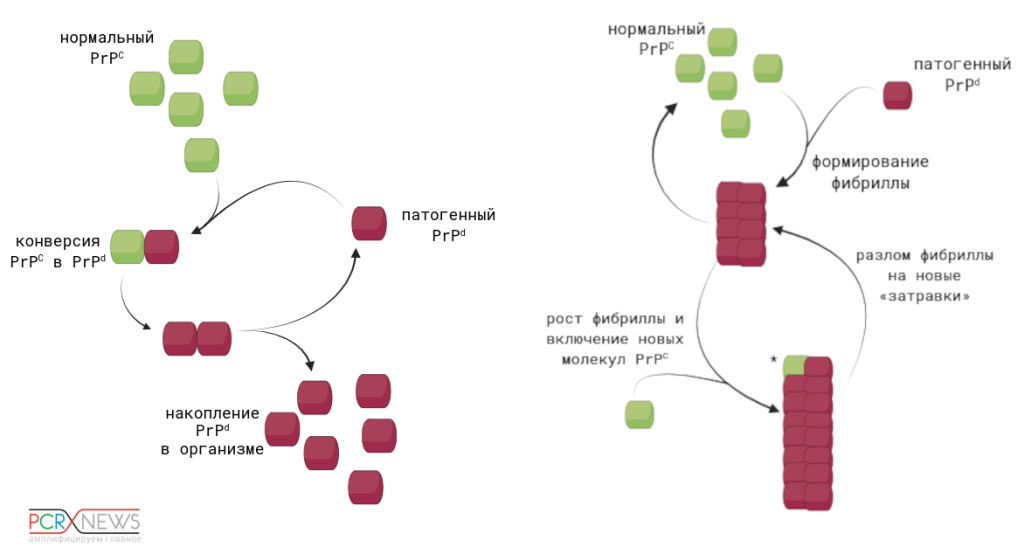 Две основные гипотезы того, как прион «портит» нормальный белок: гетеродимерная (слева) предполагает, что отдельная молекула патогенной изоформы PrPd связывается с молекулой нормального прионного белка PrPС и запускает изменение ее конформации в еще одну патогенную молекулу. Фибриллярная гипотеза (слева) отличается тем, что прионы (PrPd) существуют в клетке только в форме фибриллы. Когда нормальный белок связывается с ее концом, он приобретает патогенную укладку и становится частью фибриллы. Изображение создано с помощью BioRender.com
Две основные гипотезы того, как прион «портит» нормальный белок: гетеродимерная (слева) предполагает, что отдельная молекула патогенной изоформы PrPd связывается с молекулой нормального прионного белка PrPС и запускает изменение ее конформации в еще одну патогенную молекулу. Фибриллярная гипотеза (слева) отличается тем, что прионы (PrPd) существуют в клетке только в форме фибриллы. Когда нормальный белок связывается с ее концом, он приобретает патогенную укладку и становится частью фибриллы. Изображение создано с помощью BioRender.com
Накопление прионов губительно для клеток и тканей. Наиболее уязвима нервная ткань, в первую очередь головной мозг. Остановить накопление или разрушить уже сформировавшиеся молекулы PrPd крайне затруднительно. Все известные на сегодняшний день прионные болезни человека представляют собой различные формы нейродегенераций, они считаются неизлечимыми и смертельными.
Как можно заболеть (и, главное, как не заболеть) прионными болезнями?
Для ответа на этот вопрос нужно знать основные пути передачи прионов человеку. Их три: первый и основной из них — поглощение аномальной формы приона с пищей. Заражение возможно из-за того, что, как уже говорилось, PrPd крайне устойчивы к расщеплению протеазами (в том числе к ферментам желудочного сока).
Хорошая новость — чаще всего для распространения инфекции необходимо, чтобы первичная последовательность патогенного и «своего» белка организма совпадала, поэтому передача прионов редко бывает межвидовой. По этой же причине один из факторов риска заражения прионами — это каннибализм.
Наиболее известный пример связанного с каннибализмом прионного заболевания человека — болезнь куру, смертельная нейродегенерация, которая встречается у аборигенов племени форе в Папуа-Новой Гвинее. Члены этого племени практиковали ритуальный каннибализм — во время поминального пиршества они съедали части тела умершего родственника. Заболевание распространялось в первую очередь среди женщин и детей, поскольку мальчики старше 6–8 и взрослые мужчины практически не участвовали в таком обряде и никогда не ели мозг, содержание прионов в котором особенно высоко. (Ситуация, впрочем, осложняется долгим инкубационным периодом болезни куру — он может превышать 50 лет).
С этим связан интересный случай недавнего отбора в человеческой популяции. У представителей племени форе, переживших эпидемию куру, описаны два защитных варианта в гене PRNP. Один из них приводит к замене аминокислоты в белке PrP —глицин в положении 127 аминокислотной цепочки заменяется на валин (G127V). Такой вариант обнаружен исключительно у людей из племени форе. Он встречается у половины женщин, проживающих на этой территории, и не встречается у больных куру или любых других группах населения. Анализ родословных, проведенный в 2009 году авторами статьи в NEJM, выявил значительно меньшую заболеваемость куру в семьях — носителях варианта G127V. По-видимому, этот вариант обеспечивает некоторую защиту от прионных заболеваний.
Инфекционный прион может попасть в организм и другими путями, например, на нестерильных хирургических инструментах. Есть данные, что передача прионов может происходить воздушно-капельным путем.
Другой путь — наследственный — связан с мутациями в гене PRNP, который кодирует основной прионный белок человека. В этом гене описано более 20 мутаций, приводящих к развитию наследственных прионных болезней, например, фатальной семейной бессоннице или болезни Крейтцфельдта-Якоба (еще раз подчеркнем: она бывает как наследственной, так и инфекционной, то есть может развиться и при появлении «собственных» прионов, и при заражении «внешним» прионом). Все такие мутации наследуются аутосомно-доминантно, то есть достаточно одной копии мутантного гена — болезнь проявится, даже если вторая копия гена будет нормальной.
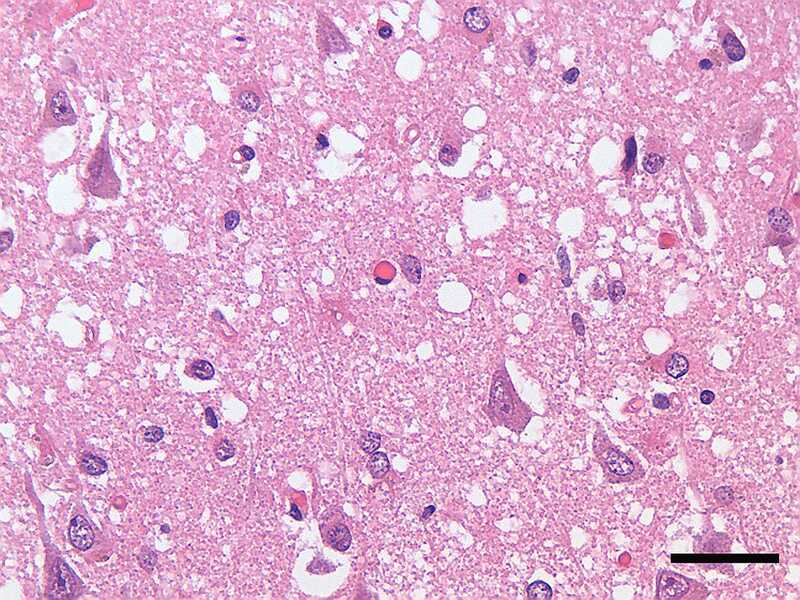 Нейродегенерация, вызванная прионами, на микрофотографии среза коры головного мозга человека, умершего от болезни Крейтцфельда—Якоба.
Нейродегенерация, вызванная прионами, на микрофотографии среза коры головного мозга человека, умершего от болезни Крейтцфельда—Якоба.Credit:
commons.wikimedia.org Tulemo | CC BY-SA 4.0
Наконец, самый загадочный случай: спорадическое, или спонтанное развитие прионной болезни. Нормальный белок PrPC по каким-то причинам (например, из-за мутации, возникшей в отдельной клетке, или модификаций после синтеза) сворачивается в патологическую конформацию.
Но гарантированной защиты от прионных заболеваний пока что не существует.
Неужели совсем нет способа выявить опасность, пока не стало совсем поздно?
Способы обнаружения прионов существуют. Ученые уже придумали лабораторные методы, которые в теории можно было бы применять не только для пищевой промышленности, но и для диагностики у людей.
Один из вариантов — использовать тот же механизм, по которому прион «портит» нормальные белки. Такую же реакцию можно провести в растворе с нормальным прионным белком, добавив в него белок из исследуемого образца. Если там присутствовала патогенная форма, она запустит сворачивание всех остальных молекул в неправильную конформацию, и это изменение можно детектировать.
Другой способ — иммуноферментный анализ. Так называют лабораторное исследование, при котором для обнаружения конкретного белка и/или измерения его количество в образце используют антитела со специальными метками.
Однако перечисленные методы — это отдельные лабораторные анализы. В теории их можно было бы применять в пищевой промышленности, однако сертифицированных тестов для проверки мяса на прионы в России нет.
А при выявлении продукты пришлось бы уничтожить. Патогенная изоформа приона — это крайне стабильный белок, который устойчив к действию ультрафиолета, ультразвука, проникающей радиации и кипячению. Кроме того, даже при сжигании образца в течение 10 минут в нем может сохраниться инфекционная активность приона, если температура горения недостаточно высока. Обычная бытовая термообработка — скажем, жарка мяса, — тем более не поможет. (Кипячение молока тоже, однако насчет его опасности нет и подтвержденных данных — аномальные прионы в молоке не выявляли.)
Так что методы, все же способные уничтожить прионы в пище, почти наверняка сделают ее непригодной к употреблению. Проще и надежнее изъять и сжечь.
Как диагностируют болезнь Крейтцфельдта-Якоба у живых людей, без биопсии мозга?
Диагностика начинается с неврологических симптомов. Есть целый список клинических критериев, по которым можно предположить болезнь Крейтцфельдта—Якоба. Прежде всего, врач должен заподозрить ее в случае любой быстро прогрессирующей деменции, которая сопровождается определенными неврологическими проявлениями.
Помимо самой деменции, к ним относятся нарушение координации, непроизвольные сокращения мышц, дрожь (тремор) конечностей или паралич. Также важные признаки, на которые врач должен обратить внимание, — это ослабление силы мышц, нарушения зрения и иногда галлюцинации.
Для получения более подробной картины пациента, у которого подозревают болезнь Крейтцфельдта—Якоба, могут направить на магнитно-резонансную или позитронную эмиссионную томографию (МРТ или ПЭТ) головного мозга. С их помощью можно выявить характерные сигналы, например, признаки атрофии участков мозга или нарушений метаболизма в нем. МРТ обычно информативнее в таких случаях, чем ПЭТ.
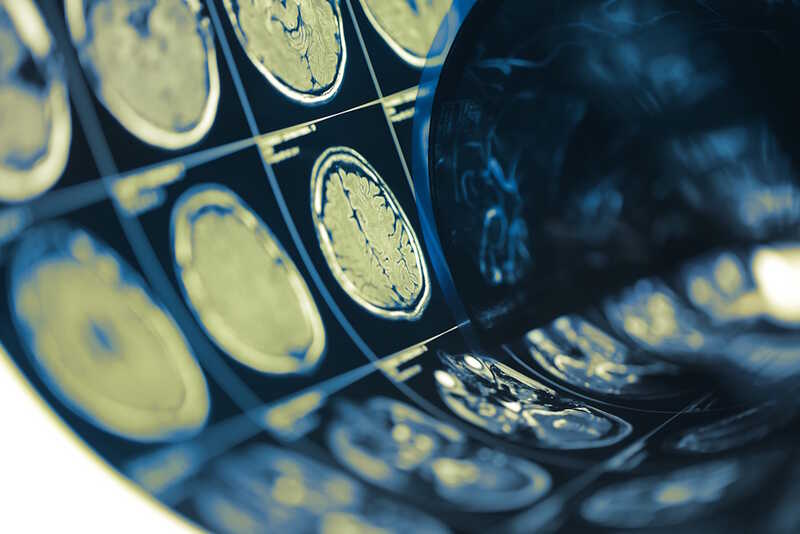 Credit:
Credit:123rf.com
Наконец, существуют методы лабораторной диагностики, с помощью которых можно выявить патологическую изоформу приона в организме. Обычно у пациента берут образец спинномозговой жидкости (СМЖ), который анализируют на неспецифические биомаркеры быстрой нейродегенерации и на сам прион. Для выявления приона проводят так называемый тест на конверсию в реальном времени. Он покажет, есть ли в СМЖ прион, способный запустить превращение нормального прионного белка в патогенный. Чувствительность такого теста, то есть вероятность правильно выявить болезнь, составляет от 92 до 97,2%.
В редких случаях на лабораторный тест все-таки отправляют образец биопсии мозга, взятый прижизненно. Однако это крайняя мера, и для проведения теста нужно отдельное согласие пациента или его родственников, а сам анализ менее чувствителен, чем лабораторное тестирование СМЖ — от 20 до 60%. Проводят его только в том случае, если все предыдущие исследования не дали однозначного результата и причина симптомов у больного все еще неясна.
Впрочем, несмотря на возможность диагностики прионных болезней, пока не найдено способа их лечения.
Прионные болезни считаются неизлечимыми. А может, все-таки можно их вылечить?
Ученые пытаются найти способ. В 2001 году доктор Прузинер, открывший прионы, выдвинул гипотезу, что большинство нейродегенеративных заболеваний связано с накоплением аномального белка в пораженных областях головного мозга. Следовательно, для борьбы с заболеваниями необходимо остановить накопление патогенной изоформы и устранить уже свернувшиеся в нее молекулы белка. Предложено несколько подходов, как это сделать.
1. Активная иммунотерапия — перспективно, но опасно
Один из способов — вызвать иммунный ответ на PrPd. Но он затруднен аутотолерантностью — собственный белок, пусть и патогенный, и измененный, не вызывает иммунного ответа. Долгое время изучались стратегии преодоления толерантности организма к своему прионному белку. Похожий метод предлагали для лечения болезни Альцгеймера — исследователи пытались вызвать иммунный ответ на амилоидные бляшки, которые накапливаются в мозге пациентов наподобие того, как это происходит с прионами. Однако первое клиническое испытание такой терапии выявило недопустимый уровень токсичности — у пациентов развивался менингоэнцефалит.
2. Моноклональные антитела — признаки успеха обнаружили после вскрытия
Введение моноклональных антител к прионному белку продемонстрировало обнадеживающие результаты в рамках клинических исследований. Опыты на клетках и животных подтвердили, что они подавляют переход PrPC в аномальную форму, а в 2022 году провели первое КИ болезни Крейтцфельдта–Якоба. Шесть пациентов получили моноклональное антитело к клеточному прионному белку (PRN100). Несмотря на терапию, всем больным пациентам становилось хуже, неврологические симптомы прогрессировали. Впрочем, впоследствии результаты вскрытия головного мозга двух пациентов не выявили признаков нейротоксичности и позволили предположить, что PRN100 может способствовать удалению PrPSc.
3. Направленная иммуномодуляция — безрезультатно
Прионы, как и раковые опухоли, легко уклоняются от иммунной системы, поэтому некоторые коллективы ученых предложили использовать те же подходы, что и для усиления иммунного ответа на рак — иммунотерапию. В этом контексте тестировалась блокировка PD-1 — одна из распространенных иммунотерапий, усиливающих иммунный ответ, которая улучшила исходы лечения у многих онкопациентов. Однако она не повлияла на накопление прионов и течение прионной болезни.
4. Генная терапия — многообещающе, но до людей еще не дошли
Терапии на основе антисмысловых олигонуклеотидов (АСО) уже одобрены для лечения таких наследственных патологий, как спинальная мышечная атрофия (известный и в России препарат Спинраза) и мышечная дистрофия Дюшенна. АСО — это препараты, которые «вмешиваются» в продукцию белка, например, мешают выработке патогенной формы или способствуют выработке нормальной формы. Наследственные прионные болезни, связанные с мутациями PRNP, составляют около 15% всех прионных заболеваний человека, поэтому АСО предложили применять и для них.
Еще один подход, основанный на идее вмешаться в экспрессию прионного белка, — это РНК-интерференция. Как и терапия прионных заболеваний с помощью АСО, она нацелена на снижение уровня патогенной формы PrPd за счет уменьшения общей экспрессии PrPC.
Кроме того, в 2025 году ученые точечно отредактировали геном зараженных прионами мышей — прямо в их организме — и показали, что это замедляет накопление патогенного белка. Мыши, которым исправили ген прионного белка, прожили дольше.
Генную терапию прионных болезней протестировали на клеточных и животных моделях, подтвердив, что она подавляет накопление PrPSc и замедляет прогрессию прионных болезней. Однако оценку безопасности для человека и клинические испытания эффективности методов еще не проводили.
5. Химиотерапия — не самый популярный вариант
Чтобы заблокировать распространение приона, предлагалось также получить химиотерапевтические агенты, которые будут нарушать связывание PrPSc с непатогенной формой прионного белка или блокировать изменение конформации. На эту роль предлагали поликатионные или полианионные соединения, но подход не получил широкого распространения.
6. Направленная деградация белков — есть свои трудности
Конечно, напрашивается предложение: если для клетки губительны отложения прионов, эти отложения нужно разрушать, чтобы спасти больного. Для этого можно применять направленную деградацию белков, однако терапии, основанные на ней, сталкиваются с проблемами. К ним относятся недостаточная растворимость или, в случае лечения нейродегенераций, способность проникать через гематоэнцефалический барьер, а также метаболическая нестабильность самих молекул-разрушителей или их токсичность. Возможность направленной деградации прионов подтвердили на мышах, и метод считается перспективным, поэтому его пытаются оптимизировать для лечения людей.
7. Стволовые клетки — а что, если начать с восстановления погибших нейронов?
Как и в случае других нейродегенеративных заболеваний, один из возможных способов борьбы с последствиями токсичности прионов — это восстановление поврежденных участков мозга клеточными технологиями. Рассматривается получение из стволовых клеток новых нейронов для замены поврежденных прионами, однако пока этот вариант исследуется не очень активно.
Устойчивость к прионным инфекциям возможна?
Вместо ответа «да» на этот вопрос вспомним уже упомянутых каннибалов племени форе. Возможность генетической устойчивости доказана на них. Кроме того, известно, что от прионных болезней защищены собаки. Анализ генетических вариантов, специфичных для собак, выявил две ключевых замены — Asn104Gly и Ser107Asn — влияющие на конформационный переход основного прионного белка и его стабильность.
Очевидно, что прионными болезнями неспособны заразиться виды, у которых отсутствует собственный прионный белок.
Всегда ли прионы таят в себе опасность?
Нет. Описан как минимум один яркий пример организмов, у которых прионы (в том числе их агрегаты), судя по всему, лишены патогенной роли. И это
грибы.
Впервые грибные прионы были описаны у пекарских дрожжей Saccharomyces cerevisiae. На них же была показана регуляторная роль прионов, и, что самое интересное, прионы грибов служат примером истинно цитоплазматического наследования — передачи информации, независимой от генома. Как и инфекционные агенты у животных, прионы грибов способны передаваться от клетки к клетке, но вместо губительного воздействия они обеспечивают перенос информации.
 Credit:
Credit:123rf.com
Кроме того, белки с прионными свойствами были описаны у арабидопсиса (Arabidopsis thaliana) — популярного модельного растения.
Если у меня нет мутации, мне можно не бояться?
Даже если не брать случаи спорадических и наследственных прионных болезней, заражение порой все еще происходит, порой совершенно неожиданно.
В июне 2019 года французская исследовательница Эмили Жомен скончалась в возрасте 33 лет от болезни Крейтцфельдта—Якоба. Это произошло спустя 10 лет после того, как во время эксперимента с мышами, инфицированными прионами, она порезала палец. После этого пять исследовательских институтов Франции ввели трехмесячный мораторий на исследования прионов.
Еще одна печальная история произошла во Франции в конце 2000-х годов. Детям с задержкой роста вводили экстракт гипофиза (отдела мозга) крупного рогатого скота, рассчитывая, что содержащиеся в нем вещества ускорят рост маленьких пациентов. Однако экстракт содержал патогенную форму прионов. В результате более 200 детей заболели болезнью Крейтцфельдта—Якоба, той самой новой формой этой болезни, которой человек может заразиться от коров.
Похожая ситуация происходила в Великобритании, однако в этом случае использовали гормон роста человека, полученный из тканей умерших людей. С 1959 по 1985 год более 1800 человек получили такой гормон, из них около 80 впоследствии заболели болезнью Крейтцфельдта—Якоба. Аналогичные заражения из-за гормона роста подобного происхождения наблюдались и в других странах, после чего препарат был изъят из обращения.
Однако в данном случае все не ограничилось именно этой прионной болезнью. Оказалось, что пять человек, которые получали такое лекарство, в возрасте 40–50 лет стали страдать от симптомов, похожих на проявления болезни Альцгеймера. Ученые, которые провели подробный анализ этого случая, пришли к выводу, что болезнь Альцгеймера, вероятно, может передаваться наподобие прионных инфекций.
Боязнь прионов — одна из причин, по которым российская вакцина «Спутник V» против коронавирусной инфекции долго не получала одобрение в странах Европы. Регуляторные органы требовали предоставить документацию о происхождении фетальной бычьей сыворотки, опасаясь передачи коровьего бешенства. Такие требования возникли после уже упомянутой вспышки в 1980-х годах; опасения подпитываются тем, что случаи заражения людей болезнью Крейтцфельдта—Якоба происходили до недавнего времени (считается, что последний — в 2016 году).
Такие труднопредсказуемые случаи, а также отсроченное проявление болезни — через многие годы после попадания в организм приона, и отсутствие способов лечения не внушают оптимизм. Опасения по поводу прионов не утихают.
Утешить может лишь статистика: прионные заболевания крайне редки, причиной смерти они становятся в единичных случаях на миллионы.
В 34 странах, по которым имелась статистика случаев прионных болезней с 1993 по 2020 год, суммарно было зарегистрировано 27 872 случая. Россия в этот список стран не входит — официальных данных по заболеваемости прионными болезнями в публичном доступе нет.
Основная доля прионных болезней в мире приходится на спорадическую форму болезни Крейтцфельдта—Якоба (24 623 случаев). Также в мире зарегистрировано 485 случаев заражений из-за врачебной ошибки, о которых говорилось ранее, и 232 случая заражений от крупного рогатого скота. При этом наблюдается тенденция к увеличению как числа случаев прионных болезней в год, так и смертности от них. Однако она сильно различается между конкретными странами. Наибольшее число случаев зарегистрировано в США, Франции, Германии, Италии, Китае, Великобритании, Испании и Канаде (в порядке убывания).




 0
0