Меню
Меню





 Все темы
Все темы
Как без потерь масштабировать транскриптомику единичных клеток
Изучение экспрессии генов в единичных клетках позволяет установить, как локальные изменения влияют на биологию человека. Однако существующие методы анализа не справляются с большими объемами данных, что приводит к противоречивым выводам. Ученые из США представили CSI-GEP — новый подход к машинному обучению, который de novo выявляет транскрипционные программы, сопоставляя их с типами клеток и активностью биологических процессов в них.
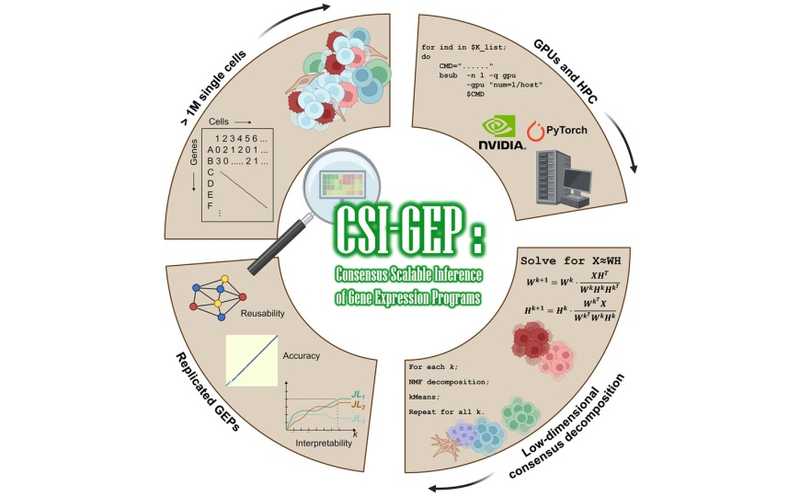
Новый алгоритм обработки данных scRNA-seq использует для вычислений мощности графического процессора, благодаря чему работает в десятки раз быстрее некоторых существующих аналогов, не уступая им в точности.
Credit:
Cell Genomics (2025). DOI:
10.1016/j.xgen.2024.100739 |
CC BY-NC-ND
Секвенирование РНК единичных клеток (scRNA-seq), появившись около 15 лет назад, обеспечило прорыв в биологических исследованиях, позволив обнаружить, например, гетерогенность клеток в мозге, особенности раковых клеток с лекарственной устойчивостью, а также новые типы и функции иммунных клеток. Однако трудности с воспроизведением и масштабированием анализа сдерживают дальнейший прогресс. Опубликованная в Cell Genomics статья предлагает возможное решение этой проблемы — алгоритм машинного обучения совместил нестандартный взгляд на обработку данных и крайне высокую производительность.
Анализ данных scRNA-seq, как правило, опирается на жесткую кластеризацию в двумерных проекциях (например, UMAP). Однако такие методы могут сильно искажать данные и требуют настройки большого количества параметров. Эти данные также можно моделировать как недискретные «программы экспрессии генов» (gene expression programs, GEP) — такие подходы позволяют сохранить структуру данных, однако плохо масштабируются и не имеют устоявшегося метода для выбора ключевых параметров. Ученые из США разработали новый подход — «консенсусное и масштабируемое вычисление программ экспрессии генов» (CSI-GEP) — и показали, что он позволяет воспроизводить GEP как на реальных, так и на смоделированных данных с высокой достоверностью и производительностью.
Метод основан на машинном обучении без учителя. Алгоритм выполняет неотрицательное матричное разложение на независимых выборках из общего набора данных, повторяя его при разных предполагаемых значениях параметра k. Затем программы экспрессии, выявленные при нескольких различных разложениях, кластеризуются с помощью k-средних, причем идентификация GEP по этим кластерам также основана на независимых выборках из общего набора. В то время как все существующие методы предполагают, что существует оптимальное значение k, которое позволяет восстановить k значимых GEP, CSI-GEP делает концептуально иное предположение: в данных существует истинный набор GEP, а модель будет стремиться восстановить эти GEP в диапазоне значений k, одновременно определяя ошибочные GEP, соответствующие шуму. Последнее достигается с помощью итеративного разложения — оно позволит отсеивать невоспроизводимые программы экспрессии, то есть шум, и оставлять те, которые соответствуют реальным биологическим особенностям данных. Эта стратегия требует подгонки модели при значениях k, которые значительно больше, чем количество истинных GEP в данных.
Самым затратным с точки зрения вычислительных мощностей является многократное матричное разложение. Авторы работы проверили несколько доступных реализаций и выбрали среди них torchMNF — в отличие от других методов, эту имплементацию можно выполнять на графических процессорах. При минимальном среди всех вариантов времени работы (8 часов, тогда как максимальная длительность вычисления у одной из реализаций превысила 780 часов) torchMNF показал сопоставимую точность, которую оценивали на заранее смоделированных данных scRNA-seq с известными характеристиками.
Эффективность CSI-GEP оценивали в сравнении с тремя уже существующими алгоритмами определения программ экспрессии по данным scRNA-seq: iNMF, SignatureAnalyzer-GPU (SA-GPU) и single-cell variational inference (scVI). Для анализа использовали смоделированный набор данных из миллиона клеток, включавший 40 специфичных для типа клеток программ экспрессии (около 25 000 клеток каждого типа) и еще один, общий для четырех типов. Оказалось, что только CSI-GEP смог корректно восстановить все GEP без ложноположительных и ложноотрицательных результатов: iNMF недооценил их размерность и выдал 25 GEP, а два других алгоритма «перестарались» и получили слишком большую размерность данных, включив в выдачу ложноположительные результаты и разделив некоторые исходные GEP на несколько более мелких.
CSI-GEP также превзошел другие методы при исследовании реальных данных. С его помощью анализировали не известные заранее данные двумя способами: в одном случае проверяли способность алгоритма выявлять GEP, обогащенные известными клеточными маркерами, а в другом определяли, насколько хорошо он определяет техническую погрешность в данных. Проверку проводили на данных scRNA-seq нейробластомы, к которым добавили один образец нормальной ткани. Как и на смоделированных данных, CSI-GEP показал во всех экспериментах более высокую производительность и точность — он обнаруживал в соответствующих GEP большее количество известных маркерных генов ожидаемых типов клеток (иммунных, опухолевых, эндотелиальных и т.д.).
CSI-GEP позволяет работать и с крупномасштабными наборами данных, недоступными для многих других методов. Его применили к клеточному атласу целого мозга мыши, включающего 2,2 миллиона единичных клеток из 13 различных участков. Алгоритм выявил 483 программы экспрессии генов — намного больше, чем обычно описывают в исследованиях с использованием scRNA-seq. Сопоставление этих GEP со списками известных маркерных генов показало, что в целом выявленные программы экспрессии отражают типичные характеристики клеток мозжечка и моторной коры. Также возможности CSI-GEP продемонстрировали на атласах раковых опухолей — их часто критикуют за несоответствие первичным опухолям, хотя в целом результаты работы с ними противоречивы. Прямое сравнение гетерогенности экспрессии в опухолях человека и в соответствующих клеточных линиях, лежащих в основе атласов, провели именно с помощью CSI-GEP. Оказалось, что большинство опухолевых линий все-таки сохраняют преобладающие паттерны экспрессии первичных раковых опухолей, однако встречаются и исключения.
Таким образом, новый алгоритм может найти широкое применение в работе с крупномасштабными наборами данных scRNA-seq, чтобы накапливать новые, биологически значимые данные и выводы. Сам GSI-SEP лежит в открытом доступе на GitHub.
Источник
Liu, Xueying et al. CSI-GEP: A GPU-based unsupervised machine learning approach for recovering gene expression programs in atlas-scale single-cell RNA-seq data. // Cell Genomics, Volume 5, Issue 1, 100739 (2025). DOI: 10.1016/j.xgen.2024.100739
Вам будет интересно
 100
100
 0
0