
 Меню
Меню





 Все темы
Все темы
МД-2023: Сателлит компании «Альбиоген». Современный рынок NGS: вызовы и решения
Сателлитный симпозиум компании «Альбиоген» «NGS — вызовы современного рынка» прошел в несколько нестандартном ключе. Вместо презентации брендов или производителей фокус аудитории был переключен на сам предмет, интерес к которому и объединил присутствующих. Обсуждались фундаментальные понятия в области секвенирования нового поколения, и современные методов и решений, присутствующих на отечественном рынке.

Александр Лавров
Фото:
Татьяна Сашина
В первом докладе Оксана Рыжкова, зав. УКП ФГБНУ «Медико-генетический научный центр им. Н.П. Бочкова», проследила путь NGS в диагностике — от «диковинки» до инструмента для национального скрининга. Прошло совсем немного времени с того момента, как секвенировали геном человека (2003 год), и на рынке появились первые NGS-секвенаторы (2005 год), а примерно 10 лет назад появились и решения для клинической диагностики. Этот тип диагностики развивался очень быстро. С использованием NGS стало возможным изучать не только фенотип, но и генотип в привязке к нему. Появился вал работ по открытию новых генов, новых форм заболеваний и по генной терапии. Оксана рассказала, как в 2015 году они с коллегами разрабатывали собственную систему диагностики методом NGS для детекции точечных мутаций в гене DMD. Тогда же появилась такая проблема, как ошибки в интерпретации результатов NGS, которые не были понятны ни врачам, ни лабораторным генетикам. Остро встал вопрос о формировании инструкций по работе с данными NGS в клинике, и в 2017 году появились первые российские методические рекомендации, которые впоследствии претерпели обновление.

Одним из первых этапов развития NGS-диагностики было создание различных таргетных панелей генов. Позднее проводились исследования информативности таких панелей, и сложилось мнение, что большие панели генов секвенировать не имеет смысла: если предполагается работа с какой-то патологией, где необходима обширная дифференциальная диагностика, например, миопатии, то лучшим вариантом будет сразу секвенировать экзом. Панели генов — хороший метод диагностики, но в случае заболеваний, для которых не нужна обширная дифференциальная диагностика (например, фенилкетонурия).
Следующим этапом стало создание такой таргетной панели, как клинический экзом (CES). C одной стороны, секвенирование полного экзома в тот момент (2018 год) было финансово сложным, а с другой — данных по корреляциям фенотипов и генотипов известно немного. Поэтому сотрудники МГНЦ разработали собственный метод секвенирования клинического экзома, который до сих пор является достаточно информативным. Несмотря на это, в настоящее время исследователи переходят к изучению полного экзома (WES).
Секвенируя полные экзомы, специалисты МГНЦ смогли определить, для каких групп заболеваний результаты оправдывают выбор данной методики. Например, для патологии ЖКТ и печени информативность диагностики будет около 20 %, для заболеваний сердечно-сосудистой системы — 30 %, а для заболеваний почек — уже 40 %. Эти результаты сходятся с мировыми данными. Таким образом, секвенирование полного экзома —информативный метод диагностики, однако более чем в половине случаев он пока что не дает результатов.
Второй находкой от работы с полными экзомами стало обнаружение в изученной специалистами выборке случаев повторяющихся, считавшихся ранее ультраредкими, заболеваний, которые, как оказалось, распространены в РФ шире, чем в других странах Теперь врачи их знают и направляют пациентов на исследование нужного им гена, тем самым сокращая финансовые и временные затраты на постановку диагноза.
Далее в работу МГНЦ было внедрено полногеномное секвенирование (WGS). В настоящее время получено уже больше тысячи геномов. Специалисты сравнили информативность полноэкзомного и полногеномного секвенирования и сделали вывод, что первый подход почти не уступает второму. Разница в информативности составляет от 2 до 6 %, что сходится с литературными данными. Однако в геноме есть много регуляторных областей, о которых мы пока ничего не знаем, информация о них еще только начинает накапливаться. С этой стороны геномное секвенирование имеет преимущество, так как дает большую возможность для переанализа данных.
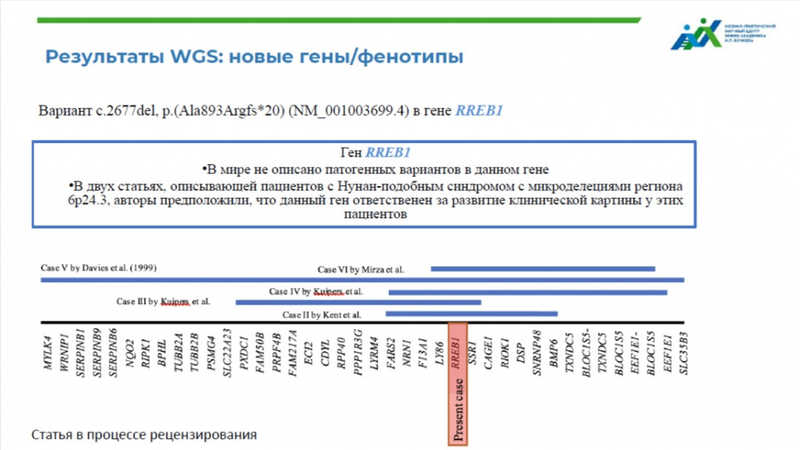
Геномное секвенирование дает специалистам возможность выявлять новые гены и новые фенотипы. В качестве примера докладчица привела случай пациента, направленного с диагнозом «задержка психомоторного развития». На киническом осмотре врачи МГНЦ поставили диагноз «заболевания из группы РАСопатий», но секвенирование не выявило патогенных вариантов в генах, описанных как ассоциированные с моногенной патологией. Однако был выявлен loss-of-function вариант в гене RREB1. В мире не было описано патогенных вариантов в данном гене, но в двух статьях упоминались пациенты с Нунан-подобным синдромом, у которых были выявлены CNV (протяженные делеции), затрагивающие область генома, в котором расположен этот ген. Был сделан вывод, что точечные мутации в этом гене RREB1 ассоциированы с Нунан-подобным фенотипом. Так специалисты МГНЦ описали новый ген, статья о нем находится в процессе рецензирования.
С 2023 года в России проводится неонатальный скрининг на 36 заболеваний. Одним из этапов диагностики первичных иммунодефицитов (ПИД) является экзомное секвенирование. Ранее специалисты МГНЦ планировали ввести диагностику с помощью панели генов и изучили имеющуюся литературу, а также данные о структуре и патологиях по разным популяциям. Они выделили несколько генов (IL2RG, JAK3, ADA, RAG1 и другие), ассоциированных с ПИД. Однако впоследствии стратегия поменялась, и пациентам провели экзомное секвенирование. В результате часть генов совпала, но у большинства больных были обнаружены гены, которые специалисты не ожидали увидеть (IGLL1, NBN). В мире известно всего четыре пациента с патогенными вариантами в гене IGLL1, а по данным МГНЦ их уже более 10. Это самый частый ген, патогенные варианты в котором приводят к агаммаглобулинемии. Это очень позитивная информация, так как подходы к лечению этого заболевания известны, детям дают антитела, и им не нужна пересадка костного мозга.
Таким образом, в настоящее время полногеномное секвенирование внедряется в практику даже на уровне скрининга, и буквально за 8-10 лет произошел переворот в диагностике наследственной патологии. Есть основания надеяться, что внедрение длинных прочтений и искусственного интеллекта сделает диагностику орфанных заболеваний еще более информативной.
Александр Лавров, ведущий научный сотрудник лаборатории редактирования генома МГНЦ, прочитал лекцию о теоретических основах секвенирования нового поколения.

По словам докладчика, в основе любой платформы лежат три ключевых этапа: создание ДНК-библиотеки, секвенирование как таковое и первичный анализ данных. На старте подготовки ДНК для анализа стандартной методикой является таргетное обогащение путем гибридизации, когда из расщепленной ДНК с помощью зондов выделяются интересующие нас фрагменты, либо путем амплификации с использованием праймеров.
Затем на основе обогащенного либо случайно расщепленного генома происходит создание библиотеки. Библиотека представляет собой набор фрагментов ДНК с присоединенными адаптерами. Адаптеры содержат последовательности для связывания с подложкой, на которой происходит секвенирование, праймеры для начала секвенирования и баркоды для различения образцов.
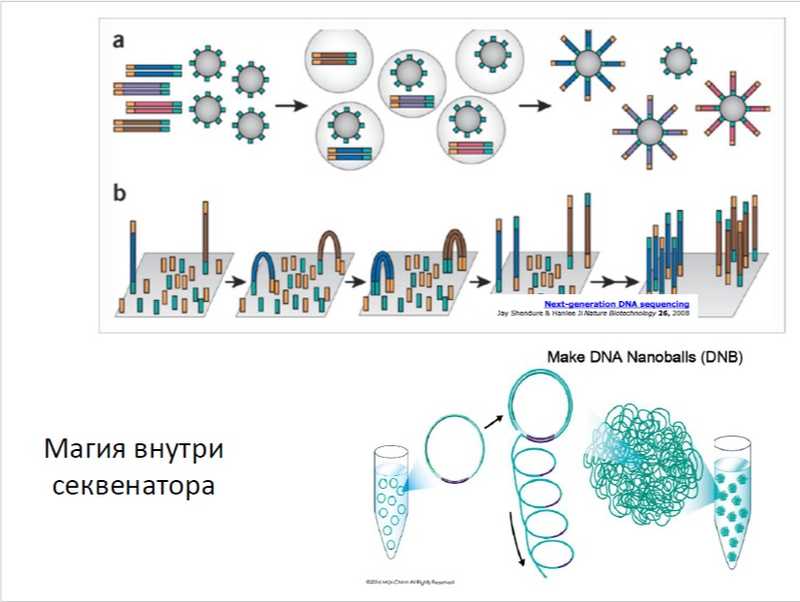
Есть три технологии секвенирования, которые в настоящее время наиболее широко используются в России. Технология Illumina реализуется на проточных ячейках. Они представляют собой два стекла на небольшом расстоянии друг от друга, которые поделены на отдельные линии, чтобы можно было загружать образцы разного типа. Библиотека садится на подложку, обработанную полиакриламидом; внесение эмпирически рассчитывают таким образом, чтобы предварительно денатурированные молекулы располагались на расстоянии друг от друга. Затем происходит запатентованная компанией Illumina мостиковая ПЦР (bridge PCR), которая позволяет образовать кластер идентичных молекул вокруг изначально одноцепочечной молекулы, случайно попавшей в это место. После этого начинается секвенирование: к праймеру добавляются терминирующие флюоресцентно меченные нуклеотиды. Сначала чтение происходит с одного конца молекулы, затем с другого, в другом направлении, и получаются парные риды.
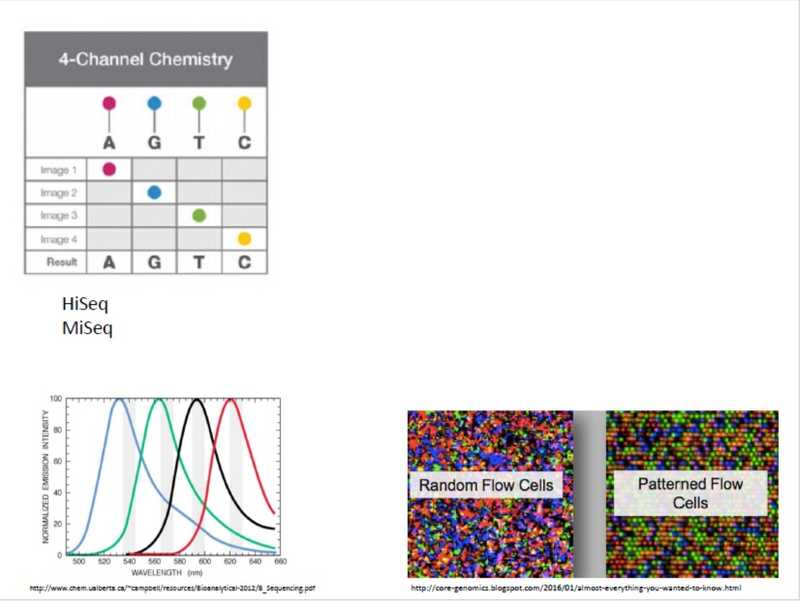
Изначально технология использовала четырехцветную химию — аналогично секвенированию по Сэнгеру, реализованную в приборах HiSeq и MiSeq. Но затем количество используемых флуорофоров стало уменьшаться для снижения стоимости секвенирования. Так, в приборах MiniSeq, NextSeq и NovaSeq используется двухцветная химия: один нуклеотид окрашен одним цветом, другой — вторым, третий — сразу двумя, а четвертый – ни одним из них. Этот подход также позволяет сократить и количество повторов детекции сигнала. В базовом приборе iSeq, используется один флуорофор и происходит его химическая модификация между детекциями сигнала. Следует отметить, что в современных приборах применяют так называемые упорядоченные ячейки, в которых заранее задано место, куда сядет молекула. Сканировать такую ячейку прибору гораздо проще, и с нее можно получить гораздо больше информации.
В технологии компании Thermo Fisher Scientific в эмульсионной ПЦР на бусине формируется кластер из нескольких тысяч одинаковых молекул аналогично тому, что происходит на ячейке от Illumina. Далее эти бусины попадают в небольшие ячейки внутри чипа. Каждая из таких ячеек представляет собой маленький рН-метр. Поочередно добавляются немеченые нуклеотиды (это сильно удешевляет технологию). В случае присоединения нуклеотида выделяется водород, прибор фиксирует изменение рН и выдает сигнал. Александр Вячеславович отметил, что у данной технологии есть существенный недостаток — сложно различить несколько одинаковых нуклеотидов, если они располагаются подряд. Эта проблема сохраняется, несмотря на улучшения технологии анализа данных в приборе.
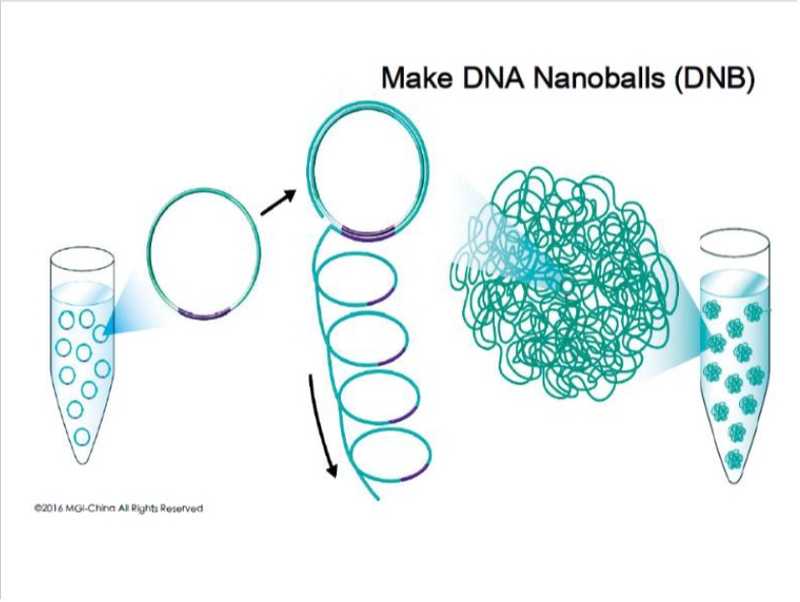
Третью технологию развивает китайская компания MGI. Библиотека с двух сторон зашивается не в линейные адаптеры, а в такие, которые могут быть закольцованы. После этого начинается амплификация по принципу катящегося кольца с высокопроцессивной полимеразой. При этом в каждом цикле происходит копирование изначальной молекулы, что снижает вероятность ошибки при амплификации библиотеки. Длинная молекула затем сворачивается в клубок (нанобол, или наношарик) и садится на подложку. Секвенирование происходит сначала в одну сторону, а затем достраивается вторая цепь: продвигаясь до предыдущей цепочки, полимераза ее вытесняет ввиду отсутствия 5’-3’ экзонуклеазной активности. В итоге получается множество идентичных молекул, синтезированных с минимумом ошибок, и второе прочтение не уступает первому по качеству.
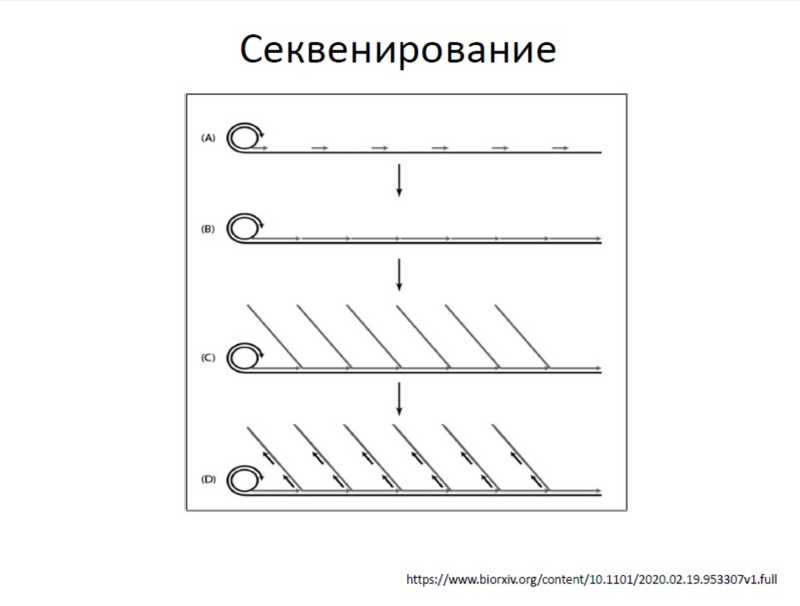
Еще одной технологией, входящей последнее время в рутинную практику, является технология длинных прочтений отOxford Nanopore Technologies. MinION —самый доступный, самый маленький, самый быстрый прибор, который можно использовать во внелабораторных условиях — по крайней мере, так утверждают маркетинговые материалы. Длинному одномолекулярному секвенированию не предшествует амплификация. Кроме того, так как секвенируется изначальная молекула ДНК, то сохраняется и вся информация о модификациях нуклеотидах, например о метилировании.
Принцип секвенирования по технологии Oxford Nanopore заключается в том, что через белковую пору протягивается молекула ДНК и измеряется проходящий через нее ток. Изменение тока зависит от того, какой фрагмент ДНК находится внутри поры. Проблема у этой технологии только одна: детектируется не один, а сразу 4–5 нуклеотидов, находящихся в поре. В последней версии пор этот момент усовершенствован — есть второй датчик, который читает еще 4–5 нуклеотидов, что позволяет получить более высокую точность. Самое удивительное, что для анализа данных уже применяются технологии ИИ: распознать сложный паттерн изменения тока и превратить его в последовательность помогают нейросети.

Еще одна технология длинных чтений реализуется компанией Pacific Bioscience. Длинный двуцепочечный фрагмент ДНК зашивается между кольцующими ее адаптерами. После этого ДНК расплетается в одноцепочечную молекулу. На ней по механизму катящегося кольца начинает работать секвеназа и с использованием флуорофоров прочитывает ее много раз. Меченые нуклеотиды детектирует датчик. Случайные ошибки статистически выравниваются, так как одна и та же молекула читается многократно.
В конце своего выступления Александр Вячеславович упомянул про секвенирование «на патефоне». Новая технология от компании Ultimagenomics — mnSBS (mostly natural sequencing-by-synthesis) — в США уже понемногу входит в клиническую практику.
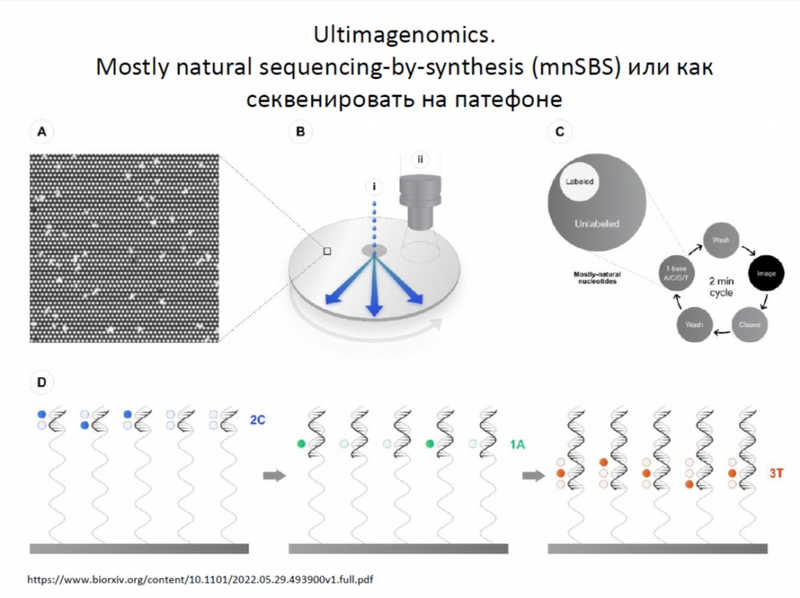
На крутящийся диск с поверхностью, состоящей из множества ячеек, сверху наносится библиотека. Это позволяет ей равномерно распространиться по поверхности. Далее происходит секвенирование с «практически натуральными» нуклеотидами — они представляют собой смесь модифицированных и немодифицированных нуклеотидов. Полученные данные анализируются с применением высокочувствительного микроскопа и нейросети. Технология очень экономична и имеет высокую скорость чтения.
Следующий докладчик, Дмитрий Полев, руководитель группы метагеномных исследований НИИ Пастера, вслед за коллегами продолжил тему обзора существующих на рынке платформ для секвенирования и, в частности, представил слушателям новое решение —секвенатор NGS Salus Pro RS (Китай).
Дмитрий напомнил, что секвенирование применяется во многих областях, в частности, для изучения геномов и метагеномов, транскриптомов и метатранскриптомов, экзомов, таргетных панелей, ампликонов, внеклеточной ДНК, ChiP-Seq и другие. Для такого множества задач необходимо и разнообразие технических решений на рынке.
В основе работы новой платформы Salus Pro лежит метод Иллюмины SBS (sequencing by synthesis) с использованием описанной выше методики мостиковой ПЦР. Амплификация происходит на проточной ячейке, построение комплементарной цепи осуществляется с использованием полимеразы и обратимой терминации флуоресцентно меченных нуклеотидов, а сигнал считывается фотосистемой с высоким разрешением. Алгоритмы обработки изображений и анализа данных запатентованы. Имеются собственные наборы реактивов разной производительности под разные задачи, разные режимы прочтений — одноконцевые (SE) и парноконцевые (PE), можно делать запуск двух ячеек одновременно. Рабочие параметры прибора аналогичны NextSeq 1000: до 1 млрд прочтений за запуск, различная длина прочтений (SE50-SE100, PE70-PE150), максимальная пропускная способность за запуск до 300 GB, время секвенирования — 9-42 часа при запуске одной ячейки и 11–45 часов при запуске двух ячеек одновременно.
Реактивы могут быть использованы по-разному в зависимости от поставленных задач, но в качестве примера производитель рекомендует одноконцевые прочтения длиной 50-100 н.п. для таргетного секвенирования, метагеномного анализа, секвенирования транскриптома, а парноконцевые прочтения — для геномного секвенирования, экзомов, транскриптомов. За год использования прибора на китайском рынке накопилось достаточное количество сведений о качестве получаемых данных, которое оказалось сопоставимым с лидером рынка. Название компании в китайском варианте переводится как «территория, отвоеванная в конкуренции», что подчеркивает ее амбициозность. Так, уже анонсирован более производительный прибор — Salus Evo (производительность до 2 ТB за запуск). Также компания развивает технологию пространственной транскриптомики и планирует интегрировать ее в свою линейку оборудования.
Мария Логачева (Центр молекулярной и клеточной биологии Сколтеха) в своем докладе прояснила место коротких чтений в мире NGS. Вначале она рассказала о том, как развивались технологии секвенирования, в качестве отправной точки взяв момент публикации генома человека в 2003 году. Бурное развитие технологий, которые дают длинные прочтения, в последние годы не означает, что короткие прочтения забыты. В конце 2022 года компания Pacific Bioscience, продвигающая именно длинные чтения, объявила, что у них теперь выходит платформа для коротких суперточных ридов.
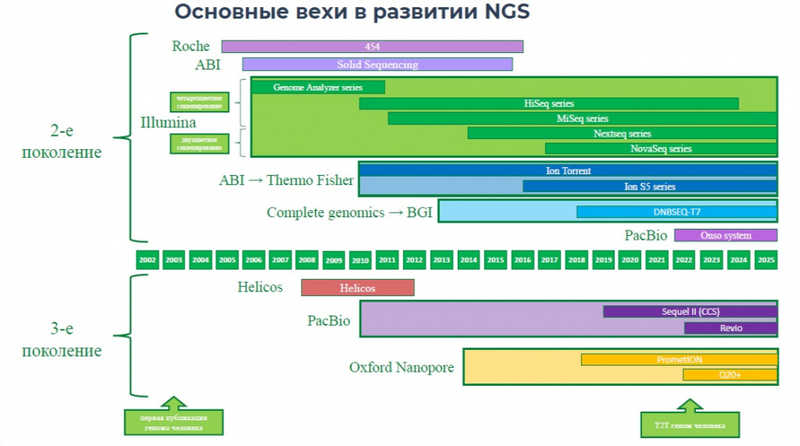
По данным NCBI абсолютный лидер по использованию в научных исследованиях — технология Illumina, однако данные по китайской платформе могут быть неточными, поскольку в Китае используют свой архив нуклеотидных последовательностей. В 2010-х годах с использованием Illumina и других платформ коротких чтений ученые проводили самые разные работы — секвенировали геномы козы, панды, огурца, анализировали метагеномы, публиковали результаты в журнале Nature, однако качество полученных данных с современной точки зрения кажется не слишком высоким. Это продолжалось до 2019–2020 годов, когда ситуация сильно изменилась: появились новые разработки от Pacific Bioscience и Oxford Nanopore, новые версии наборов для секвенирования, и произошло увеличение производительности до 200 GB с одной ячейки.
Казалось бы, сегодня, чтобы секвенировать геном de novo, можно просто выделить высокомолекулярную ДНК, прочитать на платформе Oxford Nanopore или Pacific Bioscience и собрать одним из современных сборщиков (например, Flye, Canu, Nextdenovo). Однако для того, чтобы довести сборку до уровня хромосом, очень полезно иметь данные по частотам хроматиновых контактов, которые получают с помощью Illumina, а если не хватает точности, поможет коррекция короткими прочтениями. Так, были пересобраны многие геномы. Спустя почти 20 лет после первой версии опубликована полная сборка генома человека (так называемый Т2Т-геном, от теломеры до теломеры), при этом использовались платформы PacBio и ONT для сборки, а Illumina — для верификации.
Есть задачи, для которых не важны такие преимущества платформ третьего поколения, как очень длинные чтения и отсутствия амплификации перед секвенированием. Бывают такие области NGS, в которых сами матрицы очень короткие, а амплификация необходима, так как материала недостаточно — например, в палеогеномике. Работы Сванте Паабо, получившего в 2022 году Нобелевскую премию за открытия, касающиеся геномов вымерших видов человека, сделаны с использованием коротких чтений (для расшифровки генома первого неандертальца сначала использовалась технология 454 от Roche, а последующие работы были выполнены на платформе Illumina).
Еще одно направление, для которого не нужны короткие чтения, — исследование состояния хроматина. В этом случае сначала выделяется хроматин, белки и ДНК связываются формальдегидом, затем происходит обработка ультразвуком либо нуклеазами, что позволяет избирательно расщепить ДНК в зависимости от того, насколько плотно она упакована в нуклеосомы. В результате такого расщепления получаются короткие фрагменты ДНК — несколько десятков или сотен нуклеотидов. Аналогично выполняются исследования ДНК-белковых взаимодействий, как правило методом ChIP-Seq. В онкологических исследованиях и пренатальной диагностике используется анализ внеклеточной ДНК, у которой есть две особенности, которые также делают ее хорошим субстратом для секвенирования именно короткими чтениями, — низкая концентрация и длина (около 150 п.н.).
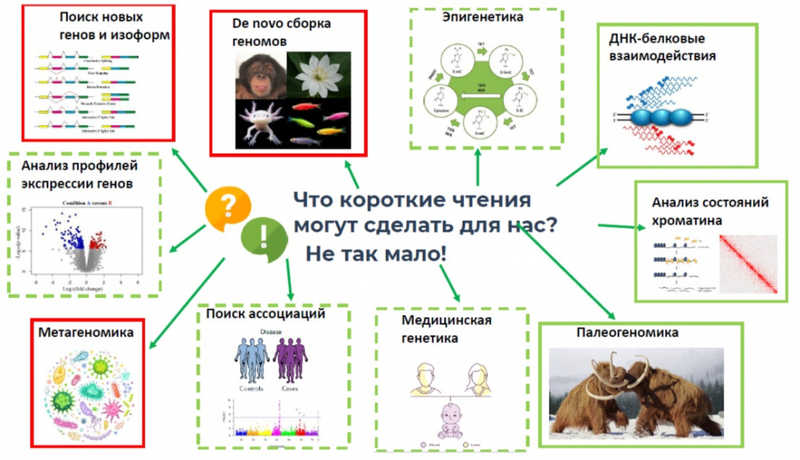
Таким образом, в настоящее время короткие чтения уже не актуальны в области поиска новых генов и изоформ, de novo сборки геномов, в метагеномике. Но они незаменимы для изучения ДНК-белковых взаимодействий, анализа состояния хроматина, в палеогеномике, и скорее всего это не изменится по мере дальнейшего развития технологий.
Информация о докладчиках
Оксана Петровна Рыжкова, к.м.н., заведующая лабораторией молекулярно-генетической диагностики ФГБНУ «Медико-генетический научный центр им. академика Н.П. Бочкова»
Александр Вячеславович Лавров, к.м.н., ведущий научный сотрудник лаборатории редактирования генома МГНЦ
Дмитрий Полев, к.б.н., старший научный сотрудник, руководитель группы метагеномных исследований НИИ эпидемиологии и микробиологии имени Пастера (Санкт-Петербург)
Мария Дмитриевна Логачева, к.б.н., старший преподаватель Центра молекулярной и клеточной биологии Сколковского института науки и технологий





 0
0










