
 Меню
Меню





 Все темы
Все темы
Новый метод NT-seq позволяет различить все три типа метилирования ДНК у прокариот
У прокариот существуют три формы метилирования ДНК, но современные способы секвенирования позволяют детектировать только одну из этих форм в одном образце. Ученые из США разработали метод, позволяющий профилировать сразу все три типа метилирования одновременно. Его опробовали на Escherichia coli и Helicobacter pylori, а также на бактериальных сообществах.
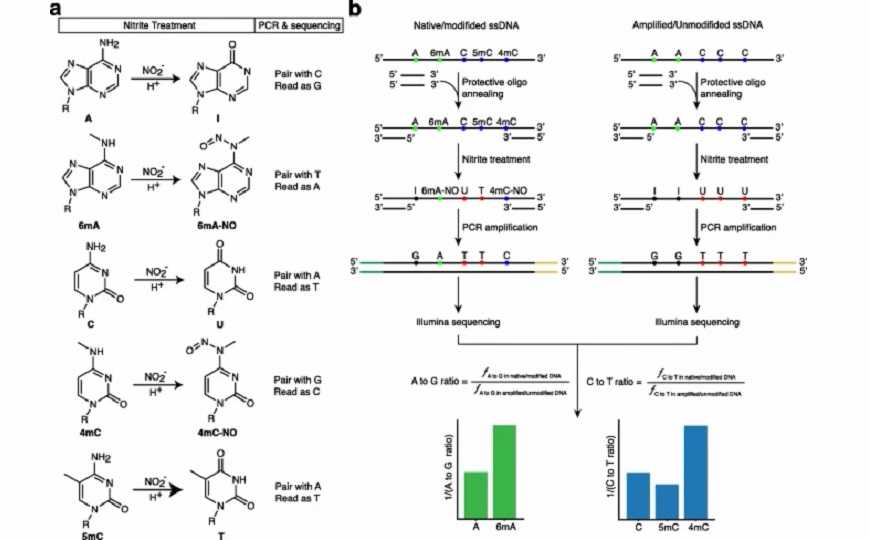
Исследователи из США опубликовали статью, в которой описали новый метод профилирования метилома под названием NT-seq, позволяющий детектировать все три типа метилирования ДНК у прокариот. Этот подход они использовали для оценки профилей метилирования в микробном сообществе и в геномах бактерий Escherichia coli и Helicobacter pylori, а также сравнили разработанный метод с уже существующими.
Большинство функциональных исследований метилирования ДНК проводится на эукариотах, но появляется все больше данных в поддержку гипотезы о существовании эпигенетических регуляторных путей у прокариот. У бактерий встречается три формы метилирования ДНК: N6-метиладенин (6mA), N4-метилцитозин (4mC) и 5-метилцитозин (5mC). Эти модификации играют важную роль в защите от вирусов, репарации ДНК и общих механизмах регуляции активности генов. Большинство методов для картирования метилирования ДНК были разработаны для 5mC, например, бисульфитное секвенирование, но у прокариот наиболее распространенной формой метилирования является 6mA. С помощью секвенирования третьего поколения SMRT-seq возможно описывать паттерны метилирования ДНК в бактериальных геномах, но для этого метода отсутствуют биоинформатические программы с открытым исходным кодом для перекрестной проверки результатов.
Существует подход к определению 6mA, аналогичный бисульфитному секвенированию. При помощи азотистой кислоты на этапе пробоподготовки происходит дезаминирование только неметилированных аденинов. Основываясь на нем, ученые разработали NT-seq. Новый метод позволяет обнаруживать паттерны метилирования всех трех типов в геномах Escherichia coli и Helicobacter pylori, а также в бактериальных сообществах.
Принцип метода основан на химической обработке образцов ДНК нитритом: NO22- индуцирует дезаминирование аденина, цитозина и 5mC, продуцируя инозин, урацил и тимин соответственно, а также нитрозилирование 6mA и 4mC с образованием нитрозилированных 6mA (6mA-NO) и 4mC (4mC-NO). После этого образец секвенирует. A читается как G, 6mA — как A, C — как T, 4mC — как C, 5mC — как T после амплификации ПЦР.
Чтобы исследовать способность NT-seq обнаруживать 6mA, авторы провели пилотные эксперименты с модифицированным олигонуклеотидом 6mA и сравнили эффективность его дезаминирования с результатом, полученным на немодифицированном олигонуклеотиде. В положении 6mA отношение A к G (отношение между частотой A к G в модифицированном/нативном образце и частотой A к G в немодифицированном/амплифицированном образце) оказалось примерно в 18 раз больше, чем в других положениях аденина. Для модификации 5mC проделали аналогичные эксперименты, и выяснилось, что отношение C к T в положении 5mC примерно на 40% выше, чем в других положениях цитозина. Это соотносится с представлением о том, что 5mC легче дезаминировать, чем цитозин.
Примечательно, что, в отличие от бисульфитного секвенирования, модификации 4mC и 5mC влияют на результат NT-seq противоположным образом, что дает возможность их различать.
Новый метод можно использовать независимо или в сочетании с другими способами профилирования метилома, например, иммунопреципитацией, что облегчит исследователям изучение роли эпигенетической регуляции у прокариот.
Источник
Li X., et al. NT-seq: a chemical-based sequencing method for genomic methylome profiling // Genome Biology volume 23, Article number: 122 (2022), DOI: 10.1186/s13059-022-02689-9

Вам будет интересно

 228
228
 0
0


 620
0
620
0
 1280
0
1280
0








