Меню
Меню





 Все темы
Все темы
Создан крупнейший каталог аллель-специфичных сайтов связывания транскрипционных факторов
Российские специалисты картировали геномные участки, замены нуклеотидов в которых влияют на связывание регуляторных белков — транскрипционных факторов, следовательно, и на активность генов. Эта база данных имеет большое значение для медицины, поскольку такие сайты часто ассоциированы с заболеваниями.
Экспрессия генов регулируется белками — транскрипционными факторами, которые обеспечивают активность тех или иных генов в разных типах клеток и на разных этапах развития организма. Участки ДНК, с которыми взаимодействуют транскрипционные факторы, которые называются сайтами связывания, — предмет особого внимания специалистов, это ключ к пониманию работы генома и механизма развития многих заболеваний.
В регуляторных участках генома, как и в кодирующих, парные хромосомы могут нести разные аллели. Замены нуклеотидов в сайте связывания влияют на его способность взаимодействовать с транскрипционным фактором. В некоторых случаях белок предпочитает определенный аллель на одной их двух парных хромосом — это явление называется аллель-специфичным связыванием. Российские ученые из Института общей генетики РАН, Института белка РАН и МФТИ разработали алгоритм для поиска сайтов аллель-специфичного связывания по всему геному и составили крупнейший каталог нуклеотидных замен, влияющих на работу транскрипционных факторов.
Для поиска и картирования сайтов связывания транскрипционных факторов применяется метод ChIP-Seq, основанный на иммунопреципитации хроматина с последующим секвенированием. В этом методе используют специфические антитела, которые прикрепляются к транскрипционным факторам по всему геному и тем самым обнаруживают места их связывания.
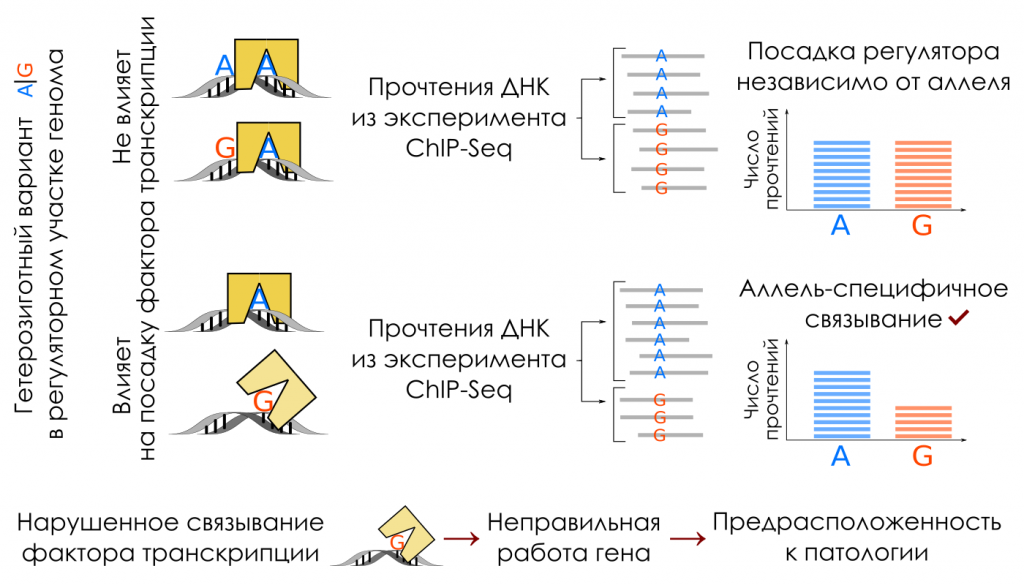 Данные ChIP-Seq позволяют отличить события аллель-специфического связывания транскрипционного фактора, то есть предпочтения определенного аллеля: об этом говорит дисбаланс числа прочтений. Credit: Пресс-релиз
Российского научного фонда
Данные ChIP-Seq позволяют отличить события аллель-специфического связывания транскрипционного фактора, то есть предпочтения определенного аллеля: об этом говорит дисбаланс числа прочтений. Credit: Пресс-релиз
Российского научного фонда
Ученые провели биоинформатический метаанализ данных, полученных в семи тысячах с лишним экспериментов, проведенных методом ChIP-Seq на разных клеточных линиях и собранных в базе GTRD; эту базу много лет разрабатывает и поддерживает новосибирский коллектив, также принявший участие в работе.
Для решения задачи нужно было преодолеть следующую проблему: во многих клеточных линиях, с которыми работают специалисты, нарушается плоидность хромосомного набора либо имеются многочисленные сегменты ДНК, присутствующие во множестве копий (copy-number variants, CNVs). В таких случаях невозможно определить, как влияет замена нуклеотида на посадку транскрипционного фактора на данную площадку, что играет роль — предпочтение того или иного аллеля или число его копий. Авторы преодолели эту сложность с помощью оригинального статистического подхода.
В чем суть этого подхода, PCR.NEWS объясняет заведующий лабораторией системной биологии и вычислительной генетики ИОГен РАН, член-корр. РАН Всеволод Макеев: «В эксперименте ChIP-Seq мы с помощью антител “вытаскиваем” сайты связывания транскрипционного фактора и можем наблюдать аллельный дисбаланс, например, 2:1, то есть одних аллелей вдвое больше, чем других. Если аллельный дисбаланс в среднем один и тот же, на протяжении довольно большого участка, то скорее всего это влияние копийности, результат того, что одного аллеля в этом районе просто больше, чем другого. Например, клеточная линия К562 — в среднем триплоидная. И если вы возьмете все аллели, которые присутствуют в геномном сиквенсе, то дисбаланс 2:1 получится потому, что на один аллель приходится больше генетического материала. Если же сильный аллельный дисбаланс возникает в каких-то отдельных сайтах, а на всем протяжении он слаб или его совсем нет, скорее всего это аллель-специфичное связывание, результат того, что транскрипционный фактор предпочитает один аллель другому».
Авторы обнаружили несколько интересных явлений. Так, с одним и тем же сайтом могут связываться два разных транскрипционных фактора (обычно в разных клеточных линиях), но эти факторы предпочитают разные аллели. «Такие случаи раньше наблюдали, но мы показали, что это явление массовое и оно может быть интересно фенотипически. Если мы возьмем уже известные варианты, ассоциированные с болезнями, то среди них это явление встречается гораздо чаще — то, что разные белки распознают разные аллели», — говорит Всеволод Макеев.
В результате ученые создали крупнейшую базу данных по аллель-специфичному связыванию ADASTRA (Allelic Dosage-corrected Allele-Specific human Transcription factor binding sites). База включает данные для 674 человеческих транскрипционных факторов (включая эпигенетические) и 337 клеточных линий. Полученная карта содержит более полумиллиона событий аллель-специфичного связывания.
Эта база данных имеет не только фундаментальное значение, но станет ценнейшим ресурсом для медицинских генетиков. Исследователи проанализировали несколько баз ассоциаций генотип-фенотип (EBI GWAS catalog, ClinVar, PheWAS, и др.) и определили, какие SNP, ассоциированные с теми или иными заболеваниями, находятся в сайтах аллель-специфичного связывания. Таким образом, эти SNP могут оказывать влияние на регуляцию работы определенных генов и обеспечивать причинную связь между генотипом и фенотипом.
«Есть еще и eQTL — локусы, связанные с уровнем экспрессии, то есть варианты, которые вызывают изменение экспрессии каких-либо генов, — добавляет Всеволод Макеев. - eQTL — это генетический термин, а мы используем молекулярно-биологический подход. С его помощью мы выяснили, что среди самых сильных eQTL очень много случаев аллель-специфичного связывания транскрипционных факторов».
Мы спросили Всеволода Макеева, связаны ли результаты этого исследования с участием его команды в глобальных проектах, таких как FANTOM и DREAM.
— Скорее нет, хотя их можно попытаться использовать, чтобы интерпретировать какие-то данные, — ответил исследователь. — Соревнование DREAM, в котором мы участвовали, больше было направлено на предсказание регуляторных участков в геноме, работающих в определенных типах клеток. FANTOM — это также не генетический проект, там нет идеи, что у вас имеются какие-то наборы вариантов, то есть индивидуальная изменчивость. Мы до сих пор никогда не подходили к этой задаче с точки зрения генетики, хотя всегда понимали, что связать индивидуальные варианты с экспрессией генов — это очень важно. Но сделать прямой анализ — до этого у нас наконец дошли руки».
Источник
Sergey Abramov, et al. Landscape of allele-specific transcription factor binding in the human genome // Nature Communications, 12, 2751 (2021); DOI: 10.1038/s41467-021-23007-0



 0
0