Меню
Меню





 Все темы
Все темы
АТФ-цитратлиаза, активируемая мутантным альфа-синуклеином, способствует его накоплению при болезни Паркинсона
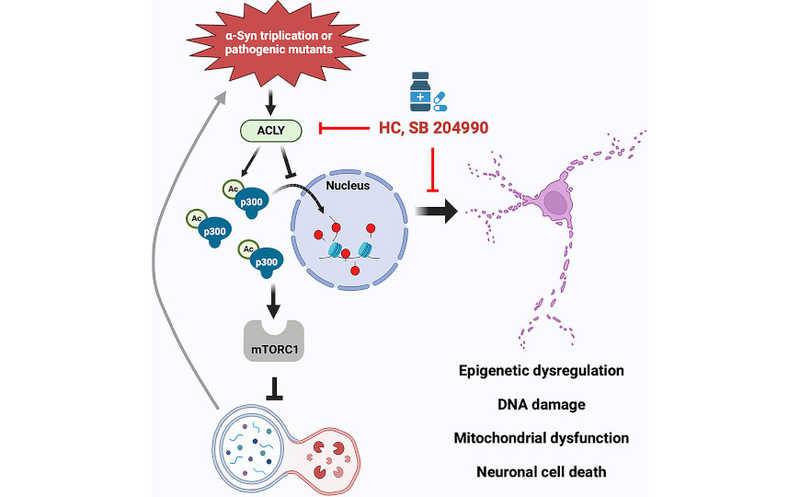
Авторы статьи в журнале Neuron охарактеризовали молекулярный механизм, связывающий мутации альфа-синуклеина (α-Syn) с нарушением аутофагии — ключевого процесса удаления патологических белков в нейронах — при болезни Паркинсона. Ученые показали, что мутация альфа-синуклеина A53T активирует фермент ATФ-цитратлиазу (ACLY), который гиперактивирует ацетилтрансферазу p300 и через нее сигнальный комплекс mTORC1, что приводит к подавлению аутофагии. Ингибиторы ACLY восстанавливали нормальную функцию аутофагии и снижали накопление патологических агрегатов α-Syn в in vitro и in vivo моделях болезни Паркинсона.
Болезнь Паркинсона — второе по распространенности нейродегенеративное заболевание, которое характеризуется двигательной дисфункцией, потерей нейронов в различных областях мозга, в первую очередь черной субстанции, и накоплением агрегатов альфа-синуклеина (α-Syn). В настоящее время не существует эффективных методов лечения болезни Паркинсона, изменяющих ее течение.
Группа под руководством ученых из Кембриджского университета установила, как мутации в гене альфа-синуклеина (SNCA), связанные с болезнью Паркинсона, приводят к сбоям в процессах аутофагии. Ключевым звеном оказался фермент АТФ-цитратлиаза, кодируемый геном ACLY. Он активируется при наличии распространенных миссенс-мутаций в SNCA (таких как A53T) и запускает каскад нарушений в работе белка p300.
Опыты на клетках нейробластомы и нейронах, полученных из индуцированных плюрипотентных стволовых клеток (ИПСК), показали, что миссенс-мутации SNCA приводят к изменению ацетилирования белков — у гистонов оно снижалось, а у цитоплазматических белков возрастало. Авторы выявили регуляторный путь, задействованный в этом — оказалось, что мутантные формы альфа-синуклеина активируют ACLY, что приводит к накоплению ацетил-КоА в цитоплазме. Это, в свою очередь, усиливает активность ацетилтрансферазы p300 в цитоплазме и активирует сигнальный путь mTORC1, который подавляет аутофагию. В результате в нейронах накапливаются токсичные агрегаты альфа-синуклеина — характерное проявление болезни Паркинсона.
Применение ингибиторов ACLY (гидроксицитрат и SB204990) возвращало p300 в ядро, нормализовало активность mTORC1 и восстанавливало аутофагию. Это приводило к снижению количества патологических агрегатов альфа-синуклеина и улучшало состояние нейронов в клеточных моделях. Результаты подтвердились in vivo — авторы провели эксперименты на трансгенных рыбках данио-рерио и мышах. Ингибиторы ACLY снижали избыточное фосфорилирование α-Syn, уменьшали активность mTORC1 и ускоряли деградацию α-Syn.
Авторы заключают, что ингибирование ACLY
может стать перспективной стратегией для лечения болезни Паркинсона, связанной с мутациями альфа-синуклеина.
Особая популяция фрагментов тРНК в крови помогает выявить болезнь Паркинсона
Источник
Son SM, et al. Alpha-synuclein mutations mislocalize cytoplasmic p300 compromising autophagy, which is rescued by ACLY inhibition. // Neuron, 2025 Apr 14:S0896-6273(25)00247-8. DOI: 10.1016/j.neuron.2025.03.028





 0
0