Меню
Меню





 Все темы
Все темы
Найдены ранее неизвестные структурные вариации генома
Новый метод определения генетических вариаций генома основан на сравнении гаплоидных генотипов человека. Используя технологии секвенирования третьего поколения, ученые поэтапно собрали 64 гаплотипа от 32 человек из 25 различных популяций. Было идентифицировано почти 108 000 структурных вариаций генома, среди них 68% новых. Это исследование не только важно для популяционных исследований, но и найдет применение в персонализированной медицине.

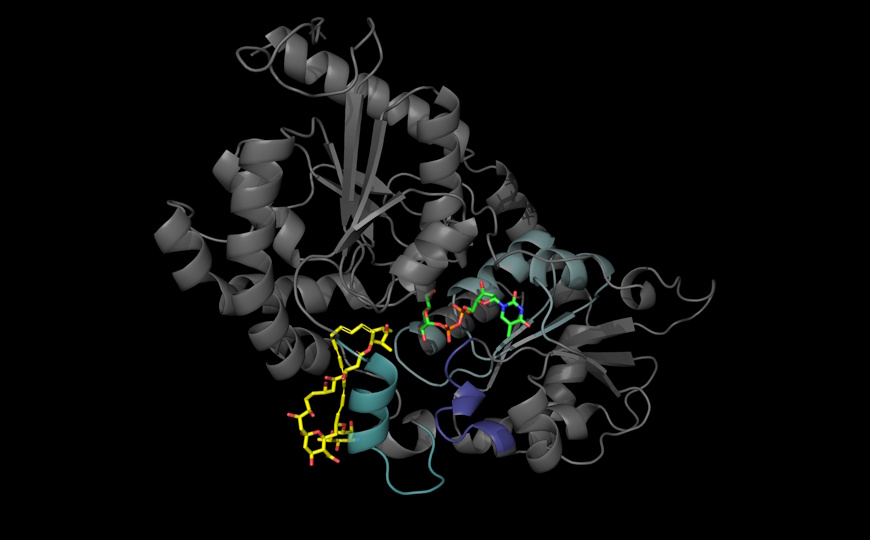
Многие человеческие геномы были собраны с помощью технологий секвенирования коротких чтений. Такие геномы трудно использовать для выявления структурных вариаций (SV) и сравнения их между отдельными людьми и популяциями. Недавние достижения в технологиях секвенирования третьего поколения, позволяющие получать длинные прочтения, значительно облегчили поиск SV – инверсий, делеций, дупликаций и длинных инсерций. Международная группа ученых пошла дальше и разработала новый метод определения широкого спектра генетических вариаций, основанный на современных технологиях секвенирования с длинными ридами.
Авторы исследования использовали одномолекулярное секвенирование в реальном времени (Pacific Biosciences) в сочетании с секвенированием одноцепочечной матрицы (Strand-seq), чтобы собрать десятки новых геномов. Путем поэтапной сборки было получено 64 набора гаплотипов от 32 человек из более чем двух десятков популяций в Африке, Северной и Южной Америке, Восточной Азии, Южной Азии и Европе. Затем путем прямого сравнения двух собранных последовательностей гаплотипов с эталонным геномом человека ученые обнаружили широкий спектр SV, включая 107 590 вариантов инсерций и делеций, более 300 вариантов инверсий и миллионы SNV, а также около 9 500 мобильных элементов.
Такой подход позволил выявить структурные варианты генома, которые не были ранее идентифицированы с применением традиционных технологий секвенирования коротких чтений. Один из авторов исследования полагает, что такие варианты, по-видимому, чрезмерно представлены у людей с определенными заболеваниями. Поэтому исследования SV будут приобретать все большее значение для диагностики.
Команда ученых также смогла обнаружить 278 горячих точек структурных вариантов в геноме человека, что дало возможность изучить механизмы их появления. Исследователи также определили более 2 100 локусов количественных признаков экспрессии, связанных со структурной изменчивостью (SV-eQTL), которые влияют на экспрессию 1526 генов.
«Благодаря новым референсным данным можно с беспрецедентной точностью изучать генетические различия на фоне глобальных генетических вариантов, что позволит получить биомедицинскую оценку генетических вариантов индивида», – подчеркивает соавтор исследования, доктор Питер Эберт.
Полученные результаты отражают генетическое разнообразие человеческих популяций и делают возможными популяционные исследования предрасположенности людей к заболеваниям.
Данные также позволяют включить весь спектр генетических вариаций генома в полногеномные ассоциативные исследования (GWAS). Это, в свою очередь, может быть использовано в персонализированной медицине и профилактике различных заболеваний.
Источник
Ebert P., et al. // Haplotype-resolved diverse human genomes and integrated analysis of structural variation // Science, 25 Feb 2021, DOI: 10.1126/science.abf7117
Цитата по EurekAlert!

Вам будет интересно


 109
109
 0
0