Меню
Меню





 Все темы
Все темы
Новая версия AlphaFold еще точнее предсказывает структуры белков и их комплексов
Новую версию AlphaFold представили 8 мая в журнале Nature. AlphaFold 3 — инструмент на базе искусственного интеллекта для предсказания структуры белков — демонстрирует более высокую точность, чем предложенные ранее аналоги и предыдущие версии AlphaFold. Разработанная модель эффективно работает с белок-белковыми взаимодействиями и модифицированными белками, а также предсказывает структуру комплексов белок-нуклеиновая кислота и белок-лиганд.
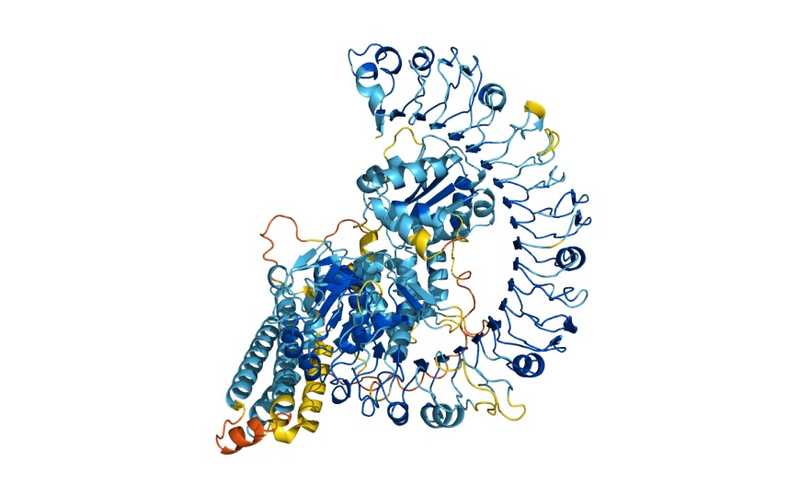
Предсказанная предыдущей версией AlphaFold структура белка арабидопсиса, предположительно обеспечивающего устойчивость к патогенам.
Credit:
alphafold.ebi.ac.uk |
CC BY
Исследователи из британских компаний Google DeepMind и Isomorphic Labs представили AlphaFold 3 — новую версию инструмента на базе искусственного интеллекта для предсказания структуры белка по его аминокислотной последовательности. По сравнению с предыдущими версиями, а также другими инструментами для предсказания структуры и взаимодействий, AlphaFold 3 демонстрирует гораздо более высокую точность в отношении взаимодействий белок-лиганд, белок-нуклеиновая кислота, антиген-антитело.
Джон Джампер, возглавляющий разработку AlphaFold в лондонском подразделении Google DeepMind, регулярно получал вопросы, способен ли этот инструмент на большее. Так, пользователи спрашивали, возможно ли предсказывать структуру белков с модификациями, которые изменяют их функции. Кроме того, исследователей, которые применяют AlphaFold в работе, интересовало предсказание структуры в комплексе с ДНК или РНК, которые нередко имеют ключевое значение для работы белка.
Общая структура AlphaFold 3 повторяет таковую предыдущей версии — AlphaFold 2 — однако в ее компоненты внесен ряд изменений. Эти изменения основаны как на необходимости обобщить работу модели на широкий спектр химических компонентов, так и на наблюдениях за работой модифицированных вариантов AlphaFold 2. Ключевой компонент AlphaFold 3 — видоизмененная версия модуля глубокого обучения Evoformer, на исходном варианте которого основывается работа AlphaFold 2. Новому варианту разработчики дали название Pairformer. Как можно догадаться из названия, он (в отличие от предыдущей версии) основан на парном представлении данных — это представление структурный модуль затем использует для генерации позиций атомов. С полученными координатами атомов работает диффузионный модуль, аналогичный тем, что используются в моделях для генерации изображений.
AlphaFold 3 может предсказывать структуры по входным данным последовательностей белков и нуклеиновых кислот, модификаций остатков и лигандов, представленных в системе SMILES. Исследователи охарактеризовали точность работы модели через среднеквадратичное отклонение положений атомов (RMSD) — уровень точности определялся как процент пар белок-лиганд с RMSD выравнивания лигандсвязывающего кармана менее 2 Å. Они показали, что AlphaFold 3 значительно превосходит классические инструменты докинга, несмотря на то, что, в отличие от них, не опирается на разрешенную структуру пары белок-лиганд (авторы учли, что такая информация может быть недоступна в реальных условиях). Методы истинного слепого докинга, например, RoseTTAFold All-Atom, также уступали новой версии AlphaFold в точности.
Также ученые продемонстрировали способность AlphaFold 3 предсказывать структуры РНК и комплексов белок-нуклеиновая кислота. Они сравнили эффективность работы своей модели с RoseTTAFold2NA и убедились, что их разработка предсказывает эти структуры с более высокой точностью. Кроме того, RoseTTAFold2NA тестировали только на структурах длиной менее тысячи остатков, тогда как AlphaFold 3 способна работать с комплексами белок-нуклеиновая кислота большей длины.
Ковалентные модификации (присоединенные лиганды, гликозилирование, модифицированные аминокислотные остатки и основания нуклеиновых кислот) также успешно предсказывались при помощи AlphaFold3, причем она работала точнее, чем AlphaFold-Multimer v2.3. Это было продемонстрировано на белок-белковых взаимодействиях и парах антиген-антитело.
Разработчики также отметили некоторые ограничения предложенной модели, связанные с особенностями стереохимии (AlphaFold 3 не всегда учитывает хиральность молекул) и неупорядоченными участками белков. При работе с последними возникают т.н. галлюцинации модели — AlphaFold 3 выдает ложные упорядоченные структуры.
Ключевой задачей молекулярной биологии, отмечают исследователи, остается понимание сложных межатомных взаимодействий в живых системах, и AlphaFold 3 делает большой шаг в этом направлении. Модель демонстрирует высокую точность предсказаний различных структур и их взаимодействий, и, хотя достижение таких результатов сопряжено со значительными трудностями, исследователи показали на примере AlphaFold 3, что создание эффективной системы глубокого обучения для этой цели возможно. Кроме того, важный результат работы состоит в улучшении предсказания взаимодействий белок-лиганд — это демонстрирует, что можно работать с широким разнообразием химического пространства, не прибегая к искусственному разделению между предсказанием структуры белка и его связывании с лигандом.
В отличие от RoseTTAFold и AlphaFold2, новая модель не будет находиться в открытом доступе — код, лежащий в основе AlphaFold3, и другая информация, полученная после ее обучения, не опубликуют. Вместо этого исследователи получат доступ к серверу AlphaFold 3, позволяющему вводить выбранную последовательность белка и интересующих лигандов.
RoseTTAFold Diffusion — новый этап в развитии белкового дизайна
Искусственный интеллект применили для создания новых редакторов генома
Источник
Abramson, J., et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. // Nature (2024). DOI: 10.1038/s41586-024-07487-w





 0
0