Меню
Меню





 Все темы
Все темы
Потеря функции белка TDP-43 вызывает характерные для болезни Гентингтона нарушения сплайсинга
Нарушения процессинга РНК меняют экспрессию генов при болезни Гентингтона. Ученые из США и Новой Зеландии обнаружили молекулярные механизмы, отвечающие за эти нарушения и связывающие болезнь Гентингтона с другими нейродегенерациями. Оказалось, что ключевую роль играет ДНК/РНК-связывающий белок TDP-43 — его неправильная локализация приводит к потере функции и изменениям сплайсинга, характерным для заболевания.
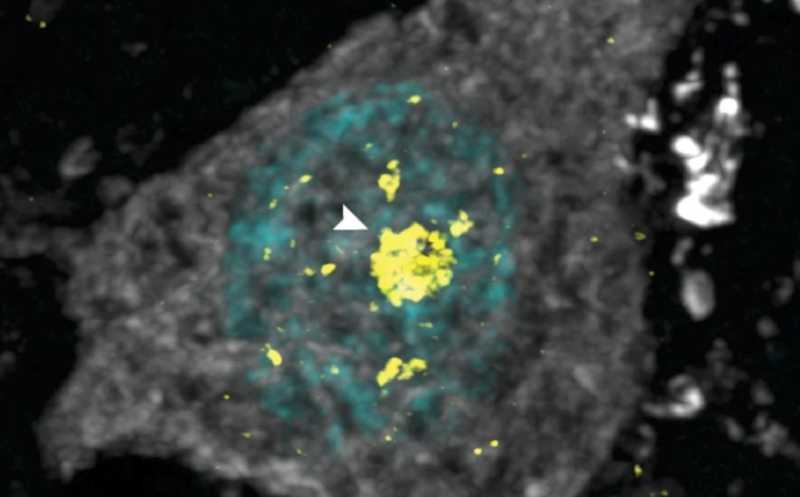
Агрегаты TDP-43 в нейроне пациента с болезнью Гентингтона.
Credit:
Nature Neuroscience (2025). DOI:
10.1038/s41593-024-01850-w |
CC BY
Болезнь Гентингтона вызывают изменения экспрессии белка гентингтина, связанное с экспансией CAG-повторов в его гене. Однако как регулируются такие изменения на уровне процессинга РНК? Исследование, недавно опубликованное в журнале Nature Neuroscience, раскрывает взаимосвязь между ключевыми регуляторами этого процесса в контексте нейродегенераций. Неправильная локализация белка TDP-43 и изменения в метилировании мРНК приводят к нарушениям процессинга РНК и ошибкам сплайсинга.
TDP-43 (TARDBP) — это ДНК/РНК-связывающий белок, необходимый для регуляции сплайсинга, который мутирует или неправильно локализуется при различных заболеваниях, в том числе боковом амиотрофическом склерозе и лобно-височной деменции. Он служит важным регулятором в процессах, связанных с нейродегенерацией. В то же время наиболее распространенной химической модификацией мРНК является метилирование аденозина по N6 положению — N6-метиладенозин (m6A) — оно изменяет локализацию, сплайсинг, стабильность мРНК и контролирует трансляцию за счет взаимодействия с РНК-связывающими белками, такими как TBP-43.
Эксперименты на мышиной модели болезни Гентингтона — линии HD R6/2 — и анализ тканей мозга пациентов с болезнью Альцгеймера и болезнью Гентингтона показали, что нарушение ядерной локализации TDP-43 и его накопление в цитоплазме меняло транскриптомные сигнатуры. (HD R6/2 — линия трансгенных мышей, экспрессирующих человеческий гентингтин с увеличенным количеством повторов (mHTT), точнее, его первый экзон.) Изменения особенно заметны в полосатом теле — области мозга, подверженной нейродегенерации при болезни Гентингтона в первую очередь.
Ученые секвенировали РНК тканей головного мозга мышей HD R6/2, чтобы выявить изменения в процессинге РНК, в том числе в сплайсинге. Самым частым нарушением оказался пропуск одного из экзонов.
С помощью моделей первичных последовательностей и их взаимодействий с белками исследователи выявили в РНК сайты связывания, ассоциированные с mHTT-зависимыми изменениями альтернативного сплайсинга и взаимодействующие с TDP-43 и метилтрансферазой 3 (METTL3), присоединяющей метильную метку к аденозину.
Выяснилось, что гентингтин с «лишними» повторами нарушает функцию TDP-43 и METTL3 в посттранскрипционной обработке их РНК-мишеней при болезни Альцгеймера. Предыдущие исследования показали, что утрата функции TDP-43 ассоциирована со снижением экспрессии генов, содержащих длинные интроны. Теперь же анализ выявил преимущественное связывание TDP-43 с кодирующими областями и 3’-нетранслируемыми регионами, а не с интронами.
Изменения в транскриптоме мышей HD R6/2 также были связаны с метилированием аденозина — обогащение m6A-сайтами было характерно для генов, экспрессия которых сильно менялась относительно контроля. Особенно это было выражено в генах, которые снижали свой уровень экспрессии.
Уже известно, что в головном мозге пациентов с болезнью Гентингтона неправильно локализуется TDP-43. В ходе этого исследования в мозге пациентов с болезнью Альцгеймера были также обнаружены агрегаты TDP-43 в цитоплазме и ядрах Map2-позитивных (зрелых) нейронов. Структуры достигали 3 мкм в диаметре и практически не детектировались в других (контрольных) образцах. METTL3 также ошибочно локализовался в цитоплазме. Полученные на пациентах данные согласовались с дисрегуляцией модификации m6A у мышиной модели болезни Гентингтона. Таким образом, между m6A-метильной модификацией РНК и TDP-43 существует связь, причем функция TDP-43 нарушается до дисрегуляции метилирования РНК.
Работа подтверждает гипотезу о том, что потеря функции TDP-43 и изменения m6A-метилирования РНК лежат в основе нарушения сплайсинга и дисрегуляции важных генов при болезни Гентингтона.
Агрегаты белка гентингтина нарушают целостность ядерной оболочки
Источник
Nguyen, T.B. et al. Aberrant splicing in Huntington’s disease accompanies disrupted TDP-43 activity and altered m6A RNA modification. // Nat Neurosci (2025). published online: 06 January 2025. DOI: 10.1038/s41593-024-01850-w





 0
0