Меню
Меню





 Все темы
Все темы
Деградацию сетчатки останавливает активация гена с помощью CRISPR-Cas
Ученые из Германии испытали на мышиной модели новый подход к генной терапии наследственной слепоты, не связанный с редактированием или доставкой функционального гена. Выполненная с помощью CRISPR-Cas активация транскрипции (трансактивация) собственного неактивного гена организма, сходного с геном, пораженным мутацией, улучшила состояние сетчатки и замедлила деградацию фоторецепторов. Работа опубликована в Science Advances.
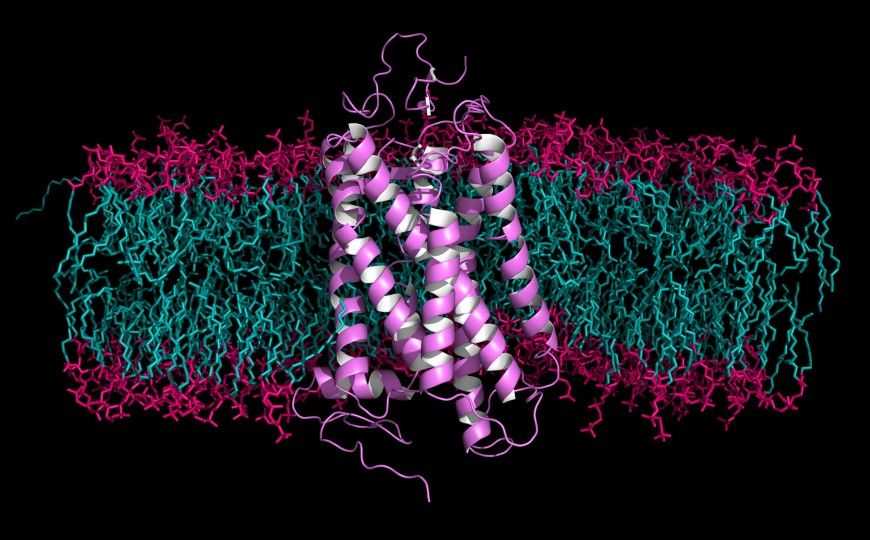
Родопсин
Credit:
petarg | 123rf.com
Один из привлекательных подходов к генной терапии — использование каталитически неактивного dCas9, слитого с активатором транскрипции (dCas9-VPR); такая конструкция, нацеленная на неактивный ген, «включает» его. У многих генов, ассоциированных с заболеваниями, есть гомологи, которые выполняют сходные функции, но экспрессируются в клетках другого типа. Активация транскрипции такого гена может скомпенсировать заболевание.
Ключевые проблемы, которые надо решить для этого, — эффективность и продолжительность экспрессия целевого гена, отсутствие побочных эффектов in vivo. Однако первая проблема — доставка dCas9-VPR в клетку. Размер последовательности, кодирующей dCas9-VPR (5,8 т.п.н.) слишком велик для упаковки рекомбинантных аденоассоциированных вирусных (rAAV) векторов, которые остаются золотым стандартом доставки генов в терапевтических целях. (Например, «самое дорогое лекарство в мире». Золгенсма — препарат на основе аденоассоциированного вирусного вектора, который доставляет в клетки человека функциональную копию гена SMN.) Ранее была показана возможность доставки расщепленной генной конструкции — в двух векторах с последующим восстановлением. Однако ее терапевтический потенциал еще не оценивался in vivo на моделях генетических заболеваний.
Исследователи из Германии применили этот подход для терапии наследственной слепоты — на мышиной модели пигментного ретинита. Это заболевание, одна из форм дистрофии сетчатки, вызывает сильное ухудшение или потерю зрения. Некоторые люди имеют симптомы с детства, у других они появляются с возрастом, но при позднем начале ухудшение идет быстрее. Болезнь вызывается мутациями в гене основного зрительного пигмента родопсина и нарушением функции фоторецепторов-палочек.
Палочки экспрессируют только родопсин, однако в колбочках большинства млекопитающих, включая мышей, экспрессируются его гомологи, опсины двух типов: коротковолновый S-опсин (ген Opn1sw) и чувствительный к средним длинам волн M-опсин (Opn1mw). Исследователи добились активации транскрипции М-опсина (Opn1mw) сначала in vitro, а затем in vivo.
Мышам с дефицитом родопсина (гетерозиготных по мутации, у которых, в отличие от гомозигот, болезнь протекает медленно) вводили субретинально в один глаз двойные rAAV векторы, экспрессирующие dCas9-VPR и гидовые РНК, нацеленные на Opn1mw, в другой глаз, для контроля, физиологический раствор. Оптическая когерентная томография, электроретинография и микроскопическое исследование спустя год показали улучшение функции сетчатки и замедление дегенерации без видимых подобных эффектов. Функция, однако, не восстановилась полностью, возможно, из-за того, что инъекция доставила препарат лишь на ограниченную площадь сетчатки.
Аналогичным образом авторы активировали in vitro еще один ген, вовлеченный в другие формы дистрофии сетчатки, — гомолог гена субъединицы управляемого циклическими нуклеотидами ионного канала, который в норме экспрессируется в колбочках. Мутации в этом гене вызывают ахроматопсию (цветовую слепоту).
Таким образом, dCas9-VPR-опосредованная активация транскрипции функционально эквивалентных генов — перспективный метод лечения генетических нарушений, отмечают авторы. Его можно использовать и для терапии генов с аутосомно-доминантным наследованием, но в этом случаех доставка активатора должна сочетаться с одновременным нокаутом мутантного аллеля.
Источник
Sybille Böhm, et al. // A gene therapy for inherited blindness using dCas9-VPR–mediated transcriptional activation. // Science Advances, 2020: 6, 34, eaba5614; DOI: 10.1126/sciadv.aba5614

Вам будет интересно
 146
146
 0
0