Меню
Меню





 Все темы
Все темы
Дисфункция митохондрий заставляет бурый жир побелеть
Исследование, опубликованное в Nature Metabolism, объясняет, как нарушение митохондриальной функции в клетках бурого жира приводит к структурной и функциональной трансформации этой ткани — бурый жир превращается в белый. Ключевую роль играет накопление D-2-гидроксиглутарата — этот метаболит усиленно вырабатывается при дисфункции митохондрий, изменяет эпигенетический ландшафт и нарушает механические свойства клеточного ядра, а в конечном счете индуцирует побеление бурых адипоцитов.

Бурая жировая ткань играет ключевую роль в несократительном термогенезе и поддержании энергетического баланса за счет богатой митохондриальной сети и экспрессии разобщающего белка UCP1. Однако при патологиях она может «белеть»: клетки утрачивают способность к термогенезу, накапливают крупные липидные капли и теряют бурый фенотип. Хотя этот процесс давно ассоциируют с митохондриальной дисфункцией, конкретные молекулярные механизмы, связывающие нарушения в митохондриях с клеточной и тканевой перестройкой, до сих пор оставались малоизученными.
Чтобы установить, как нарушения работы митохондрий влияют на судьбу бурой жировой ткани, группа ученых из Германии, Дании и Финляндии смоделировала эти процессы на мышах. Авторы использовали три линии мышей с дефицитом митохондриальной матриксной протеазы CLPP: с полным нокаутом (CLPP-KO), адипоцит-специфичным нокаутом (CLPP-AKO) и с нокаутом исключительно в бурой жировой ткани (CLPP-BKO). У всех линий наблюдалось побеление бурого жира, характеризующееся уменьшением количества липидных капель и увеличением их размера — медиана площади капель увеличилась с 500 до более чем 1,500 мкм².
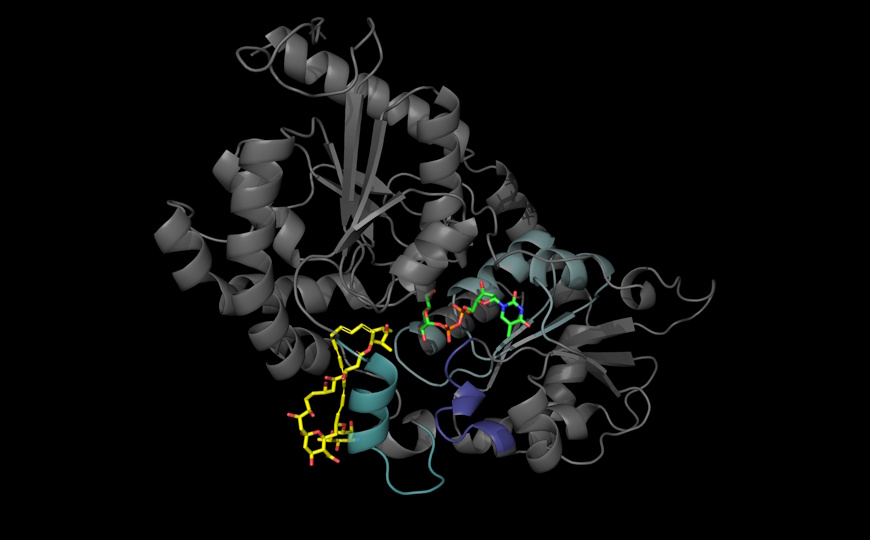
Ультраструктурный анализ с помощью электронной микроскопии выявил изменения в митохондриях: они были крупнее и содержали меньше крист. Также в них снижалось содержание комплекса I дыхательной цепи. Тем не менее, измерения потребления кислорода и скорости ацидификации среды показали, что дыхательная активность осталась стабильной — иными словами, на макроуровне функция митохондрий сохранялась. Метаболомный анализ методом масс-спектрометрии выявил значительное накопление D-2-гидроксиглутарата (D-2HG) — в жировой ткани его уровень возрастал в 2,7–3,7 раза по сравнению с контролем. Накопление D-2HG происходило только в зрелых бурых адипоцитах, но не в преадипоцитах, что говорит о зависимости процесса от дифференцировки. Источником D-2HG оказалась повышенная активность фермента фосфоглицератдегидрогеназы (PHGDH), который играет ключевую роль в синтезе серина. Его уровень вырос более чем в 5 раз. Ингибирование PHGDH с помощью малой молекулы NCT503 устраняло накопление D-2HG, уменьшало липидные капли и восстанавливало нормальную морфологию бурой жировой ткани (авторы подтвердили это гистологически).
ChIP-seq-анализ модификаций гистонов выявил, что при нокауте CLPP или после обработки D-2HG в клетках повсеместно возрастало количество H3K4me3 меток. Исследователи обнаружили 1,141 перекрывающихся эпигенетических пиков, преимущественно в промоторах генов, связанных с липогенезом и дифференцировкой — Srebp1, Cebpb, Cd36. Более 50% изменений в экспрессии генов в CLPP-дефицитных клетках соотносились с H3K4me3-ассоциированными регионами.
Одним из самых неожиданных открытий стало влияние D-2HG на механику ядра. С помощью атомно-силовой микроскопии авторы показали, что после воздействия D-2HG (при непосредственной обработке или дефиците CLPP) жесткость ядра бурых адипоцитов снижается почти вдвое. Эти изменения сопровождались деформацией ядерной оболочки и усилением контактов между митохондриями и ядрами, что подтвердилось путем 3D-моделирования. Восстановить жесткость ядер авторы смогли, ингибируя PHGDH или обрабатывая клетки холестерином — это также подчеркивает роль липидного состава в регуляции механики ядра.
Дополнительно ученые продемонстрировали, что кратковременное ингибирование митобиогенеза (например, с помощью актинона) вызывает резкое накопление D-2HG, укрупнение липидных капель и снижение ядерной жесткости даже в клетках без мутаций CLPP. Это доказывает универсальность обнаруженного механизма: митохондриальная дисфункция приводит к метаболическому сдвигу, который трансформирует эпигеном, липидный обмен и ядерную архитектуру.
Работа демонстрирует, что нарушение митохондриальной функции в бурой жировой ткани запускает каскад метаболических, эпигенетических и механических изменений, приводящих к ее побелению. Центральным медиатором этого процесса служит D-2-гидроксиглутарат, производимый PHGDH. Полученные данные ценны для понимания метаболических заболеваний и, возможно, позволят разработать методы таргетного вмешательства в побеление бурого жира.
Дефицит сиртуина 6 нарушает термогенез в буром жире и способствует ожирению
Холод заставляет бурые адипоциты переключать АТФ-синтазу в режим гидролиза
Источник

Вам будет интересно

 231
231
 0
0


 624
0
624
0
 1285
0
1285
0