Меню
Меню





 Все темы
Все темы
Эволюция в пробирке помогает определить структуру белка
Американские и канадские ученые разработали новый метод определения пространственной структуры белков, сочетающий лабораторную эволюцию и компьютерные технологии. Метод имеет ряд преимуществ перед традиционными техниками.
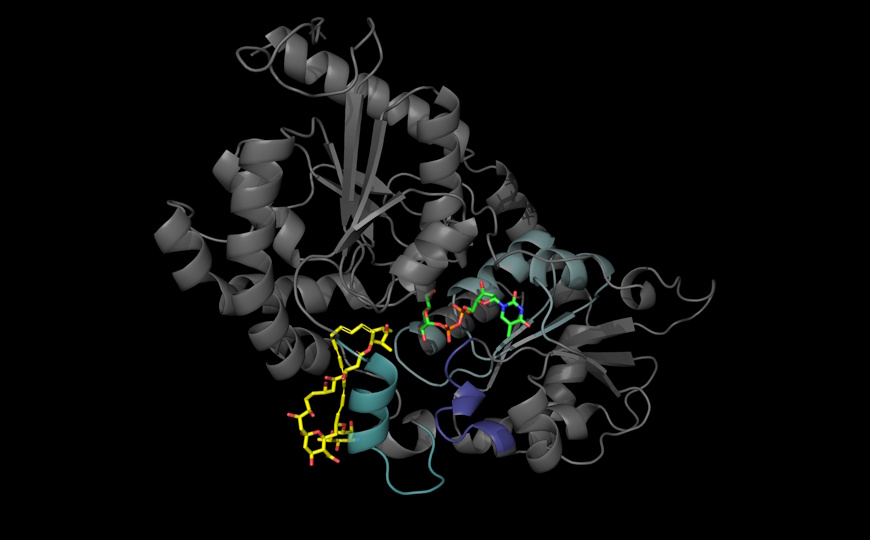
Прослеживая естественную эволюцию по изменению последовательности ДНК, можно установить, в каких частях белка происходят аминокислотные замены. Компьютерные технологии позволяют предсказать по этим данным взаимодействия между аминокислотными остатками, выделить функциональные участки у гомологичных белков и восстановить их трехмерную структуру. Авторы статьи, опубликованной в Cell Systems, предположили, что для этих целей можно использовать искусственную эволюцию в лаборатории: она происходит быстрее, чем естественная, и в контролируемых условиях.
Для проверки этой гипотезы ученые выбрали две последовательности ДНК: ген β-лактамазы PSE1, отвечающей за устойчивость к ампициллину у бактериального штамма Pseudomonas aeruginosa PSE1, и ген аминогликозид ацетилтрансферазы AAC6, обеспечивающей устойчивость к канамицину у Pseudomonas sp. ABAC61. Их подвергали случайному мутагенезу с помощью ПЦР с ошибками (error-prone PCR), в ходе которой ДНК-полимераза присоединяет 3–4% нуклеотидов некомплементарно из-за специально подобранных неоптимальных условий реакции. Чтобы отобрать функциональные варианты ферментов, библиотеки мутантных генов трансформировали электропорацией в лабораторный штамм Escherichia coli и высевали трансформанты на селективные среды с соответствующими антибиотиками (ампициллин для PSE1 и канамицин для AAS6). Неработающие ферменты не могут обеспечить выживаемость бактерий, поэтому вырастали только клоны клеток, получивших функциональный мутантный белок. Ученые провели несколько раундов мутагенеза и отбора, после чего с помощью глубокого секвенирования определили количество мутаций по сравнению с изначальными вариантами ферментов. В последовательности гена PSE1 обнаружили в среднем 34,2 аминокислотные замены (12,9% от всей длины сиквенса гена), а в AAC6 — 8,7 замен (5,9%), причем с каждым следующим циклом мутагенеза количество новых мутаций уменьшалось.
Данные обрабатывали с помощью вероятностной модели EVcouplings, которая ранее была успешно использована для идентификации коэволюционирующих паттернов в ходе естественной эволюции. Полученные карты взаимодействий аминокислотных остатков сравнили с кристаллографическими данными для предковых белков ( PDB:1G68 для PSE1 и PDB:4EVY для AAC6) из общедоступной базы данных. Ученые выявили более выраженную структурную вариабельность среди белков, полученных в ходе экспериментальной эволюции, по сравнению с естественной эволюцией. Это объясняется тем, что для эволюции в лаборатории используются сравнительно небольшие размеры популяций и условия гораздо менее разнообразные, чем в природе.
Тем не менее, авторам удалось показать, что экспериментальный эволюционный процесс в лабораторных условиях во многом похож на то, что происходит в природе, и позволяет ab initio определить взаимодействующие аминокислоты и восстановить трехмерную структуру белка в целом. Описанный метод получил название 3Dseq. Он обладает рядом преимуществ по сравнению с традиционно использующимися методами: например, 3Dseq не требует трудоемкой стадии выделения и очистки белка. Сейчас технологию совершенствуют для скрининга широкого спектра белков.




 0
0