Меню
Меню





 Все темы
Все темы
«Филоволны» отслеживают эволюцию приспособленности патогенов в реальном времени
Как меняется приспособленность патогенов и их отдельных штаммов на популяционном уровне? Чтобы отследить это, ученые из Великобритании и Франции предложили подход филоволн (phylowave), который учитывает изменения в составе популяции на филогенетических деревьях и позволяет обнаруживать линии, исходя из данных о приспособленности и эволюционных связей. Они проверили модель на наборах последовательностей респираторных вирусов и бактерий человека (SARS-CoV-2, вирус гриппа H3N2, возбудители коклюша и туберкулеза) и подтвердили, что она выявляет основные известные циркулирующие линии для каждого патогена и находит мутации, связанные с изменениями в приспособленности.
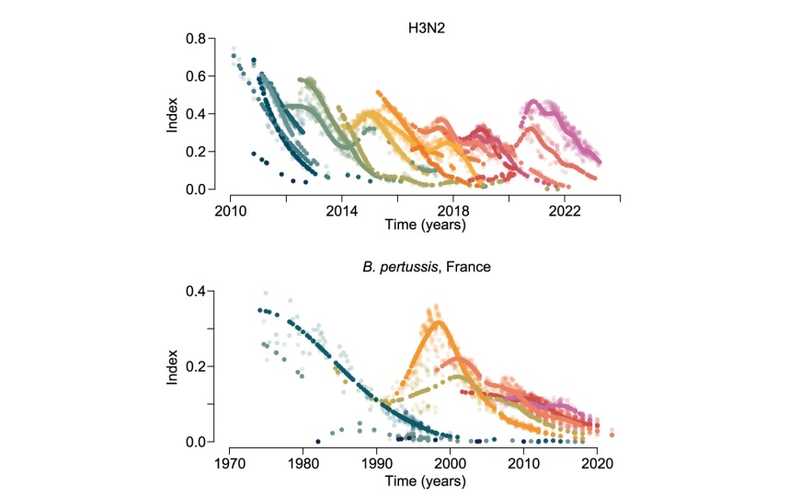
Временная динамика линий гриппа H3N2 и Bordetella pertussis — коклюшной палочки.
Credit:
Nature (2025). DOI:
10.1038/s41586-024-08309-9 |
CC BY
Состав циркулирующих среди людей штаммов у большинства патогенов постоянно меняется. Изменения окружающей среды, давление коллективного иммунитета, разная способность к инфицированию и распространению среди людей повышает жизнеспособность одних линий и приводит другие к вымиранию. Это фундаментальное свойство патогенов имеет важное значение для здравоохранения, ведь разнообразие микроорганизмов может быть связано с уклонением от вакцинного или естественного иммунитета или повышением трансмиссивности.
Один из подходов для изучения приспособленности штаммов на популяционном уровне — определение их принадлежности к известным кладам. Это могут быть линии Pango для SARS-CoV-2 или глобальные клады вируса гриппа. Приспособленность штамма оценивают с помощью моделей, которые фиксируют изменение доли отдельных линий во времени. Недостаток этого подхода — группировка штаммов по произвольным пороговым значениям аминокислотных различий или по иным соображениям ученых, не связанным напрямую с приспособленностью патогенов.
Другой стратегией является построение филогенетических деревьев. Более приспособленные штаммы будут передаваться чаще, что приведет к более высокой скорости ветвления дерева и большему количеству потомков. Для этого используют филодинамические подходы, например, модели рождения и гибели. Но такие расчеты сложно проводить из-за большого количества получаемых данных, а еще они подвержены искажениям выборки.
Ученые из Великобритании и Франции разработали новый подход, который назвали «филоволнами» (phylowave). В его основе лежит индекс, который рассчитывается с использованием генетических дистанций и измеряет успешность каждого внутреннего или терминального узла дерева с использованием заданной временной шкалы. Ученые предположили, что узлы из недавно возникшей, но более приспособленной линии будут филогенетически ближе друг к другу, чем остальная часть популяции в конкретный момент времени, так как все они будут иметь одного недавнего общего предка. Индекс позволяет динамически отслеживать появление линий и в большей степени использует информацию о недавней динамике популяции, чем данные о ее прошлой эволюции. Временную шкалу адаптируют к конкретному патогену, и ее масштаб может варьировать от месяцев (для РНК-вирусов) до лет (для бактерий).
Исследователи применили подход филоволн для моделирования эволюции четырех респираторных патогенов человека. Они взяли выборки последовательностей гемагглютинина вируса гриппа H3N2 (n = 1,476), полногеномных последовательностей SARS-CoV-2 (n = 3,129), Bordetella pertussis (возбудитель коклюша) из Франции (n = 1,248) и Mycobacterium tuberculosis из Самары, Россия (n = 998). Для каждого патогена подход выявил линии с четкими различиями в приспособленности. Например, ученые определили клады SARS-CoV-2, соответствующие вариантам альфа, бета, гамма, дельта, омикрон и другим. Линиям, выделенным с помощью подхода филоволн, близко соответствовали глобальные клады вируса гриппа H3N2 (3C.3a, 3C.2a3 и 3C.2a1b.1b), линии и сублинии M. tuberculosis и клады B. pertussis, которые определяются аллелями промотора токсина коклюша (ptxP) и фимбриального гена (fim3). Также, ученые обнаружили три новые линии B. pertussis с отчетливой динамикой индекса.
Затем ученые оценили приспособленность каждой линии в привязке ко времени. Для SARS-CoV-2 они обнаружили, что максимально приспособленной была линия 1, которая соответствует варианту омикрон XBB1.5. За ней следовали линии 5 и 7, соответствующие вариантам омикрон BA.5 и BA.1. Приспособленность линий вируса гриппа H3N2 была более однородной по всей популяции, а сами линии сохранялись в среднем в течение 3,9 лет после появления. Три линии B. pertussis (обозначенные 1, 2 и 3) появились после внедрения новой бесклеточной вакцины во Франции в 1998 году и имели самую высокую приспособленность среди всех штаммов. Это указывает на иммунное давление на динамику линий со стороны новой вакцины. Приспособленность линий Mycobacterium tuberculosis оказалась наиболее стабильной из всех четырех патогенов, что отражает давно существующую разнообразную популяцию.
Мутации, определяющие линию, присутствовали как минимум в 80% последовательностей ее представителей и не были обнаружены в предковой. Самую высокую долю таких мутаций обычно имели гены, связанные с вирулентностью. Для SARS-CoV-2 самая высокая плотность аминокислотных замен была в рецептор-связывающем домене спайк-белка, ниже ‒ в ORF1a и ORF1b, и ни одной замены не найдено в ORF10. Для B. pertussis ученые обнаружили отбор двух ранее не известных несинонимичных мутаций в гене sphB1, что предполагает конвергентную эволюцию патогена. Ген sphB1 кодирует протеазу, которая участвует во внеклеточном высвобождении нитевидного гемагглютинина — антигена бесклеточной вакцины против коклюша и ключевого фактора взаимодействия с хозяином. Для M. tuberculosis гены, связанные с устойчивостью к противомикробным препаратам, содержали самую высокую долю мутаций, определяющих линию.
В целом, подход филоволн позволяет отслеживать изменения в составе популяции патогена с течением времени в прямой связи с приспособленностью. На примере SARS-CoV-2 ученые показали, что модель способна улавливать каждую линию с задержкой в 2,2 месяца после появления, и для этого требуется всего десять последовательностей. Набор данных, использованный в исследовании, составил примерно 0,02% от всех последовательностей, доступных базе GISAID. Использование более крупного набора данных помогло бы еще больше сократить время идентификации линии.
Эпицентр возникновения высокопатогенных вирусов птичьего гриппа сместился из Азии в Европу и Африку
Источник
Noémie Lefrancq et al. Learning the fitness dynamics of pathogens from phylogenies. // Nature (2025). DOI: 10.1038/s41586-024-08309-9

Вам будет интересно
 70
70
 0
0