Меню
Меню





 Все темы
Все темы
Гистоновая ацетилтрансфераза MOF поддерживает целостность митохондрий
Немецкие ученые установили, что гистоновая ацетилтрансфераза MOF влияет на метаболизм митохондрий, ацетилируя митохондриальные белки, в частности, COX17 (часть комплекса IV). Снижение уровня MOF или COX17 приводит к фрагментации митохондрий и нарушению структуры крист. Люди с мутациями в MOF в гетерозиготе имеют признаки митохондриальных заболеваний.
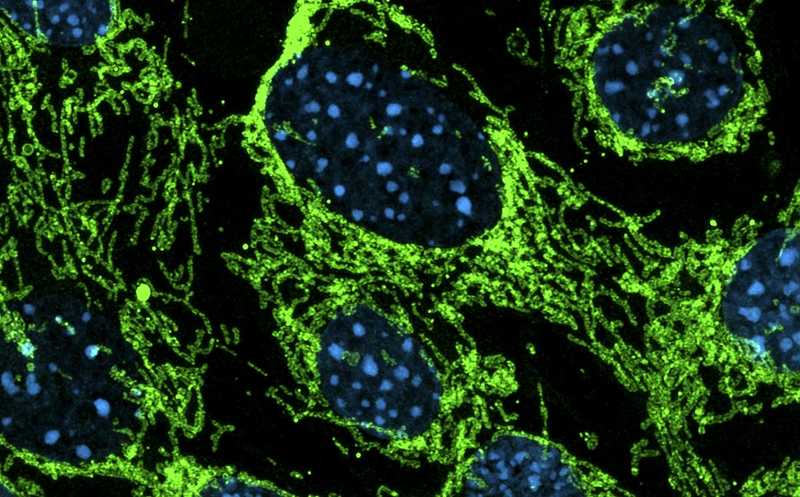
Здоровые клетки с удлиненной митохондриальной сетью.
Credit:
MPI of Immunobiology & Epigenetics, Freiburg | Пресс-релиз
Функциональная значимость комплекса гистоновой ацетилтрансферазы MOF с белками KANSL ранее была показана в нескольких исследованиях. В ядре MOF осуществляет ацетилирование гистона H4 по остатку лизина 16, что активирует транскрипцию, а отсутствие в клетке функциональных комплексов MOF-KANSL приводит к разнообразным нарушениям, в особенности со стороны митохондрий. Авторы нового исследования, опубликованного в Nature Metabolism, показали, что MOF в комплексе с KANSL ацетилирует белок COX17 (часть комплекса IV митохондрии), и это ацетилирование необходимо для нормального митохондриального дыхания в первичных клетках мыши и человека.
Используя мышиные эмбриональные фибробласты, исследователи провели серию нокаутов и показали, что отсутствие MOF, KANSL2 или KANSL3 изменяет митохондриальное дыхание. В мышиных фибробластах с нокаутами по Mof и Kansl2 активировался гликолиз, возможно, для компенсации снижения эффективности митохондриального дыхания. Кроме того, утрата MOF, KANSL2 и KANSL3 повышала уровни активных форм кислорода в митохондриях, а одновременное отсутствие MOF и KANSL2 снижало митохондриальный мембранный потенциал.
Дальнейшие исследования показали, что MOF и KANSL не только регулируют метаболизм митохондрий, но и играют важнейшую роль в поддержании их структурной целостности. Отсутствие MOF в клетках приводит к фрагментации митохондрий, кроме того, такие митохондрии почти не ветвятся, а средняя длина ветвей митохондрий и плотность крист снижаются. Тем не менее, на митохондриальной ДНК и белках, необходимых для слияния или разделения митохондрий, нокаут MOF не сказывается.
В предшествующем исследовании было показано, что в клетках HeLa MOF в комплексе с KANSL1 участвуют в контроле транскрипции компонентов дыхательной цепи, закодированных в митохондриальной ДНК. Однако на мышиных фибробластах этот результат не подтвердился, вероятно, в связи с тем, что HeLa полагается на анаэробное дыхание, а у фибробластов ведущую роль играет митохондриальное дыхание.
Наконец, авторы работы определили основную мишень MOF-KANSL в митохондриях — COX17. Ацетилирование происходит по сайтам K18 и K30. Авторы проводили нокдаун COX17 и экспрессировали в клетках мимики ацетилированного COX17 человека и неацетилируемого белка. Они показали, что ацетилирование COX17 необходимо для поддержания нормальной структуры митохондрий, причем механизм его регуляции через MOF-KANSL оказался общим для мыши и человека. Нокдаун COX17 индуцировал фрагментацию митохондрий и нарушения структуры крист — фенотип, схожий с нокаутом MOF. Восстановить нормальный фенотип мог COX17 дикого типа или миметик ацетилированного белка, но не мутант, который невозможно ацетилировать. Одновременный нокдаун MOF и COX17 не усиливал проявления патологического фенотипа. MOF-KANSL через COX17 оказывают влияние на комплекс IV, так как ацетилирование COX17 необходимо для его функции.
Авторы работы отмечают, что гетерозиготные пациенты, несущие de novo мутации в гене MOF, фенотипически близки к людям с митохондриальными заболеваниями. Для них характерны отставание в развитии, в том числе умственном, и эпилепсия. Авторы провели опыты с дермальными фибробластами контролей и людей с мутациями MOF. Исследования подтвердили, что нарушение негистонового ацетилирования может быть механизмом развития патогенного фенотипа. В клетках пациентов был снижен мембранный потенциал и нарушено митохондриальное дыхание. В то же время морфология митохондрий не была затронута. Авторы предположили, что для гетерозигот характерна неполная пенетрантность, а у клеток может быть неизвестный механизм адаптации. В любом случае, COX17 — таргет ацетилирования MOF у человека и потенциально может играть роль в патогенезе.
Исследователи предполагают, что у MOF могут быть и другие таргеты помимо COX17. Они выявляли другие гипоацетилированные митохондриальные белки в клетках с нокаутом MOF. Для проверки этого предположения необходимы дополнительные исследования.
Мутация, вызывающая болезнь Паркинсона, нарушает контакты между митохондриями и лизосомами
Источник:
Guhathakurta S., et al. COX17 acetylation via MOF–KANSL complex promotes mitochondrial integrity and function. // Nature Metabolism, 2023, DOI: 10.1038/s42255-023-00904-w
Вам будет интересно

 44
44
 0
0


 37
0
37
0