Меню
Меню





 Все темы
Все темы
Мутация, вызывающая болезнь Паркинсона, нарушает контакты между митохондриями и лизосомами
Ранняя болезнь Паркинсона чаще всего вызывается мутациями в гене белка паркина. В новом исследовании установлено, что в нейронах с мутантным паркином снижается активность белка Rab7, необходимого для контактов между митохондриями и лизосомами. Нокдаун еще одного белка, инактивирующего Rab7, нормализовал митохондриально-лизосомные контакты и аминокислотный метаболизм в нейронах, полученных из клеток человека с болезнью Паркинсона.

Болезнь Паркинсона (БП) — распространенное нейродегенеративное заболевание. Одним из его признаков на клеточном уровне является нарушение работы митохондрий и лизосом. Самая частая генетическая причина раннего развития БП — мутации в гене PARK2, который кодирует белок паркин — убиквитинлигазу, участвующую в поддержании гомеостаза митохондрий и эндолизосом. Однако каким образом паркин связан с работой этих органоидов, до сих пор не было ясно.
Авторы статьи в Science Advances установили, что при болезни Паркинсона количество контактов между митохондриями и лизосомами снижается из-за мутации гена PARK2, и это приводит к нарушению гомеостаза аминокислот. Митохондриально-лизосомальное взаимодействие, не связанное с деградацией митохондрий, открыто сравнительно недавно. Известно, что оно модулирует различные клеточные процессы, в том числе динамику органелл и обмен компонентами между ними, а при неврологических заболеваниях, включая БП, оно может быть нарушено.
Исследование проводили на дофаминергических нейронах, полученных из индуцированных плюрипотентных стволовых клеток (ИПСК) человека с болезнью Паркинсона. В качестве контроля использовали дофаминергические нейроны, выращенные из изогенных клеток (ИПСК, в которые с помощью CRISPR исправили мутацию). Транскриптомный и метаболомный анализ показали, что в этих клетках нарушен аминокислотный метаболизм и значительно изменены уровни незаменимых аминокислот изолейцина/лейцина, лизина, метионина, фенилаланина и валина, а также заменимых аминокислот аргинина, пролина и тирозина. Наблюдалось накопление аминокислот в липосомах и дефицит в митохондриях.
Авторы выдвинули гипотезу, согласно которой у людей с БП мутации гена PARK2 вызывают нарушение обмена аминокислот, так как его продукт влияет на формирование контактов между митохондриями и лизосомами. Для проверки этой гипотезы сравнили образование контактов митохондрий и лизосом в клетках HeLa с геном PARK2 дикого типа и с мутацией C431S в этом гене. В клетках, экспрессирующих паркин дикого типа, было больше контактов между митохондриями и лизосомами, чем в мутантных. В нейронах, полученных из клеток человека с БП, также наблюдалось сниженное количество контактов по сравнению с изогенными клетками. Когда нейроны человека с БП трансдуцировали геном PARK2 дикого типа, контакты между митохондриями и лизосомами восстановились. Трансдукция мутантным геном, продукт которого не способен к убиквитинилированию, такого эффекта не дала.
Известно, что митохондриально-лизосомальные контакты регулируются GTP-связывающим белком Rab7, поэтому авторы исследовали, влияет ли паркин на его функцию. Активность Rab7 в нейронах, экспрессирующих паркин дикого типа, была в два раза больше, чем в клетках, экспрессирующих мутантный паркин C431S.
Поскольку дестабилизация белка Rab7 в нейронах с мутантной формой паркина нарушала образование контактов между митохондриями и лизосомами, авторы предположили, что восстановлению контактов может способствовать нокдаун TBC1D15. Этот белок, также известный как Rab7 GAP TBC1D5, инактивирует Rab7. Действительно, нокдаун TBC1D15 увеличил количество и продолжительность контактов митохондрий с лизосомами в дофаминергических нейронах, полученных из ИПСК человека с БП, до уровней, наблюдающихся в контрольных изогенных нейронах.
Таким образом, паркин модулирует связывание митохондрий и лизосом за счет стабилизации активного, связанного с лизосомами Rab7, а нарушение регуляции контактов между митохондриями и лизосомами в дофаминергических нейронах при БП может компенсировать снижение уровня Rab7 GAP TBC1D15.
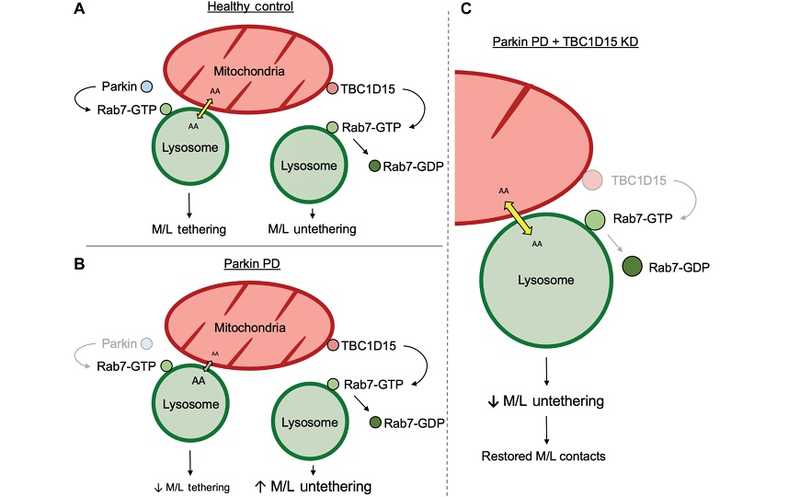 (A) В норме паркин регулирует контакт митохондрий и лизосом посредством стабилизации активного Rab7, способствуя митохондриальному и лизосомальному гомеостазу аминокислот. (B) Потеря паркина при БП снижает число контактов митохондрий и лизосом из-за потери активности Rab7. (C) Нокдаун Rab7 GAP TBC1D15 при БП восстанавливает контакты между митохондриями и лизосомами и субклеточные потоки аминокислот. Credit: Sci Adv (2023). DOI:
10.1126/sciadv.adh3347 | CC BY-NC
(A) В норме паркин регулирует контакт митохондрий и лизосом посредством стабилизации активного Rab7, способствуя митохондриальному и лизосомальному гомеостазу аминокислот. (B) Потеря паркина при БП снижает число контактов митохондрий и лизосом из-за потери активности Rab7. (C) Нокдаун Rab7 GAP TBC1D15 при БП восстанавливает контакты между митохондриями и лизосомами и субклеточные потоки аминокислот. Credit: Sci Adv (2023). DOI:
10.1126/sciadv.adh3347 | CC BY-NC
Авторы проверили, нормализуются ли аминокислотные профили нейронов и их органелл при восстановлении динамики образования контактов. Оказалось, что нокдаун гена TBC1D15 в нейронах улучшает показатели аминокислотного состава: как незаменимые, так и заменимые аминокислоты приблизились к норме. Дефицит аминокислот в митохондриях также восстанавливался после нокдауна, а характерное для БП накопление аминокислот в лизосомах было частично нормализовано.
Эти результаты подтверждают ключевую роль в регуляции контактов митохондрий и лизосом белка паркина, который поддерживает активную форму Rab7, а также указывает на то, что эти контакты необходимы для регуляции аминокислотного баланса. Нормализация характерных для болезни показателей при нокдауне TBC1D15 указывает на новые возможности для генной терапии болезни Паркинсона.
Репрограммированные нейроны из клеток кожи оказались лучшей моделью болезни Паркинсона
Источник
Wesley Peng, et al. Parkin regulates amino acid homeostasis at mitochondria-lysosome (M/L) contact sites in Parkinson’s disease. Science Advances.9,eadh3347 (2023). DOI: 10.1126/sciadv.adh3347
Вам будет интересно
 125
125
 0
0