
 Меню
Меню





 Все темы
Все темы
Полногеномная пробоподготовка Parseq Lab для Illumina, Thermo Fisher Scientific и MGI
16 мая 2022 г. российская компания Parseq Lab сообщила о запуске нового продукта в линейке Prep&Seq — базового модуля Prep&Seq™ G-fragmentation для подготовки полногеномных библиотек.

Базовый модуль Prep&Seq™ G-fragmentation подходит для широкого спектра задач. Среди них подготовка библиотек геномной ДНК, работа с продуктами ПЦР длинных фрагментов (long-range PCR, исследования микробиома, подготовка библиотек свободно циркулирующей ДНК (cfDNA). Модуль включает реагенты для ферментативной фрагментации и восстановления концов, лигирования по тупым концам и амплификации библиотек.
Ключевые свойства набора — проведение фрагментации и восстановление концов в одну стадию; подтвержденная неспецифичная активность эндонуклеазы, что обеспечивает однородное покрытие генома; совместимость с популярными в России платформами Illumina, Thermo Fisher Scientific и MGI. В зависимости от типа используемой платформы для секвенирования подбираются подходящие совместимые платформоспецифичные модули (адаптеры и праймеры). Для работы с cfDNA предусмотрен вариант протокола без фрагментации.
Входной диапазон количества ДНК — от 0,1 до 1000 нг. Длина фрагментов на выходе варьирует от 100 до 1 500 п.н. При исходном количестве ДНК не менее 100 нг возможно проведение протокола PCR-free для платформ Illumina и Thermo Fisher Scientific.
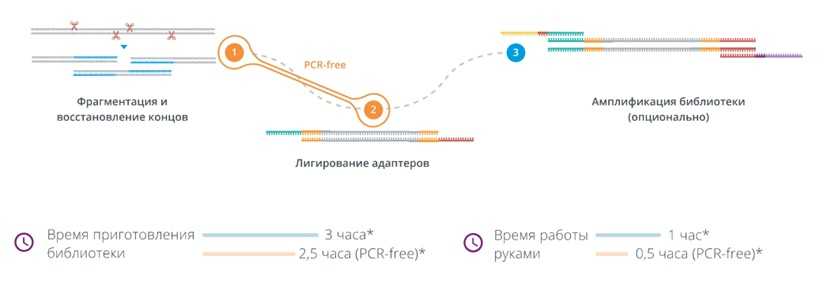 Рисунок 1. Схема полногеномной пробоподготовки с использованием модуля G-fragmentation.
Рисунок 1. Схема полногеномной пробоподготовки с использованием модуля G-fragmentation.
Средний размер фрагментов можно регулировать, изменяя объем внесенного энхансера (Рисунок 2).
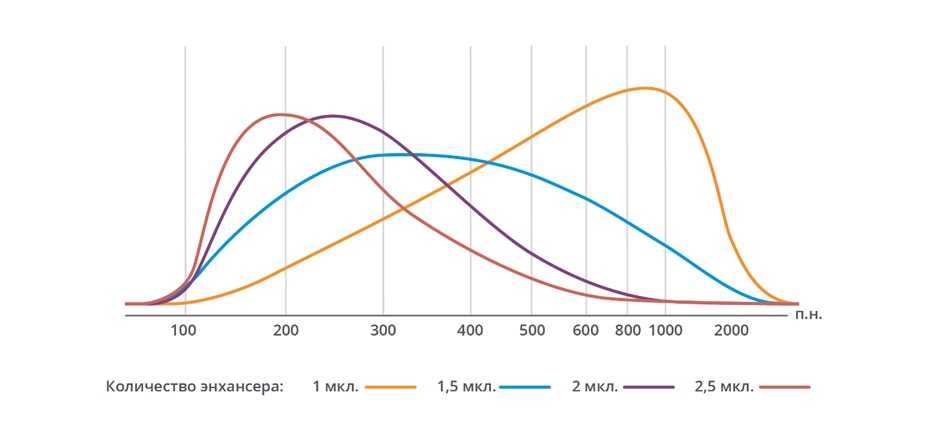 Рисунок 2. Распределение размеров фрагментов при различном количестве внесенного в реакцию энхансера. Представлены данные фрагментации геномной ДНК фага лямбда при инкубации 10 минут.
Рисунок 2. Распределение размеров фрагментов при различном количестве внесенного в реакцию энхансера. Представлены данные фрагментации геномной ДНК фага лямбда при инкубации 10 минут.
Смесь для фрагментации и восстановления не имеет выраженной мотив-специфической активности, что позволяет получать высокую однородность покрытия генома (Рисунок 3).
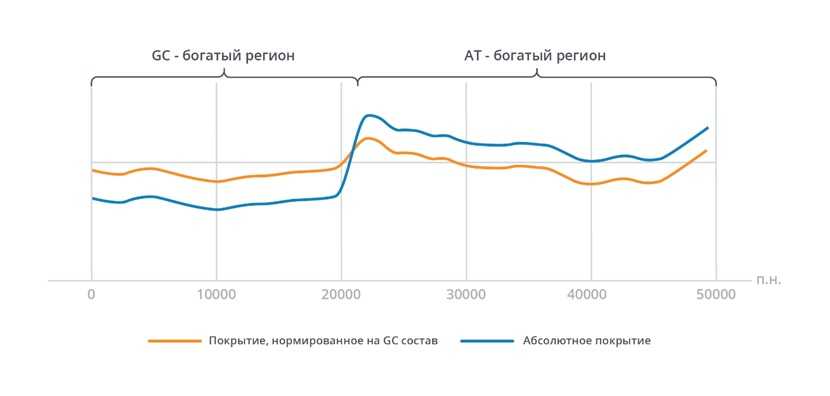 Рисунок 3. Профиль покрытия генома фага лямбда до (абсолютное покрытие) и после нормировки на GC состав (покрытие, нормированное на GC-состав). Нормализация выполнена по алгоритму Yuval Benjamini, Terence P. Speed, Summarizing and correcting the GC content bias in high-throughput sequencing, Nucleic Acids Research, Volume 40, Issue 10, 1 May 2012, Page e72.
Рисунок 3. Профиль покрытия генома фага лямбда до (абсолютное покрытие) и после нормировки на GC состав (покрытие, нормированное на GC-состав). Нормализация выполнена по алгоритму Yuval Benjamini, Terence P. Speed, Summarizing and correcting the GC content bias in high-throughput sequencing, Nucleic Acids Research, Volume 40, Issue 10, 1 May 2012, Page e72.
При использовании протокола с size selection на магнитных частицах возможен отбор более узкого диапазона размера библиотек (Рисунок 4).
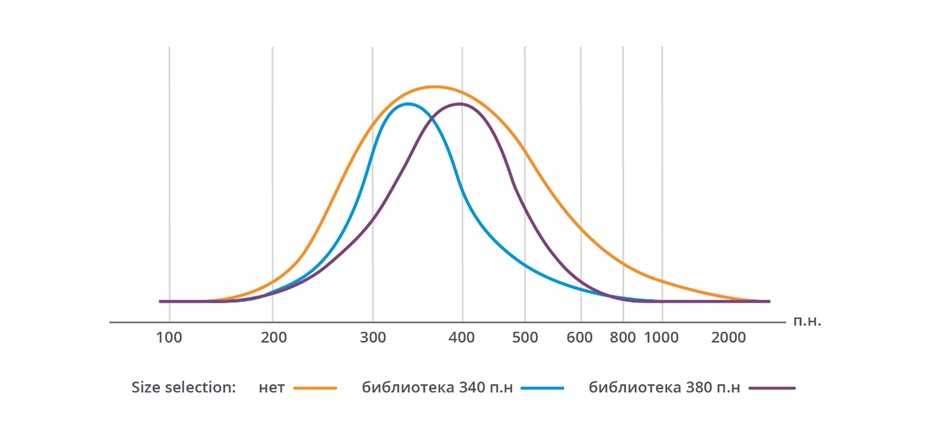 Рисунок 4. Пример изменения размеров библиотеки, полученной из геномной ДНК человека при использовании 2 мкл энхансера и 10 минутах фрагментации. Два способа очистки позволяют выделить пик целевого размера библиотеки.
Рисунок 4. Пример изменения размеров библиотеки, полученной из геномной ДНК человека при использовании 2 мкл энхансера и 10 минутах фрагментации. Два способа очистки позволяют выделить пик целевого размера библиотеки.
Ниже представлен ассортимент адаптеров и праймеров, благодаря которым можно использовать базовый модуль G-fragmentation с различными платформами для секвенирования.

Партнерский материал
Вам будет интересно




 1274
1274
 0
0










