Меню
Меню





 Все темы
Все темы
Составлена наиболее подробная структурная карта протеома SARS-CoV-2
Новый веб-ресурса Aquaria-COVID содержит подробную структурную карту протеома коронавируса SARS-CoV-2. Создатели ресурса охарактеризовали возможные структуры 27 коронавирусных белков и их связь с функциями.
Credit: Garvan Institute of Medical Research | Пресс-релиз | CC BY
За время пандемии COVID-19 ученые установили экспериментально и выложили в базу RCSB PDB (Research Collaboratory for Structural Bioinformatics Protein Data Bank, далее PDB) множество структур белков вируса SARS-CoV-2. Большая часть структурных исследований посвящена S-белку и его взаимодействиям с различными лигандами, но некоторые научные группы моделируют полный протеом (результаты одной из таких работ размещены в базе BIOZENTRUM). Для моделирования используются разные программы, среди них AlphaFold, C-I-TASSER, MODELLER, Rosetta, SWISS-MODEL. Однако для некоторых методов предсказанные структуры сильно варьируют. Кроме того, такие подходы обычно нацелены на построение модели только одного или нескольких структурных состояний для каждого белка.
Авторы новой работы поставили цель преодолеть ограничения существующих подходов и смоделировать каждое возможное состояние для каждого белка SARS-CoV-2, загруженного в PDB. Они собрали всю доступную информацию по структурам белков SARS-CoV-2 и родственных ему SARS-CoV и MERS-CoV и доработали созданную ранее платформу для молекулярной визуализации Aquaria. Новый ресурс Aquaria-COVID содержит данные о структурах белков SARS-CoV-2, пока не доступные на других ресурсах.
В работе использовали 14 белковых последовательностей из UniProt, соответствующих белкам SARS-CoV-2. В PDB с этими последовательностями связано 1 180 белковых структур. С помощью программы HHblits ученые нашли в PDB еще 880 структур в PDB, исходно относящихся к другим организмам. Их использовали как минимальные модели для белков SARS-CoV-2. Таким образом, получили 2 060 структур, покрывающих 69% протеома SARS-CoV-2. Эти структуры внедрили в Aquaria. Программа позволяет отображать структуры с подгрузкой функций UniProt, CATH, SNAP2 и PredictProtein. С помощью этих функций можно предсказывать консервативность, нарушение целостности, построение доменов, гибкость структуры, предрасположенность к мутациям и локализацию структур в инфицированных клетках. Авторы создали матричный макет, предоставляющий доступ к моделям, и структурную карту покрытия на основе коронавирусного генома, на которой суммируются результаты анализа всех 2 060 структур.
В работе описаны 27 вирусных белков, их известные и предсказанные с помощью моделирования свойства и функции, а также известные и потенциальные молекулы-партнеры. В результате сравнения модельных и экспериментальных структур ученые распределили белки по четырем функциональным группам.
Первая группа — белки, способные имитировать (mimic) человеческие белки. К этой группе отнесли три белка: NSP3, NSP13 и NSP16.
Вторая группа –– белки, которые могут блокировать (hijack) работу клеточных белков. К ним относятся NSP1, NSP3, гликопротеин S, белок оболочки и белок ORF9b.
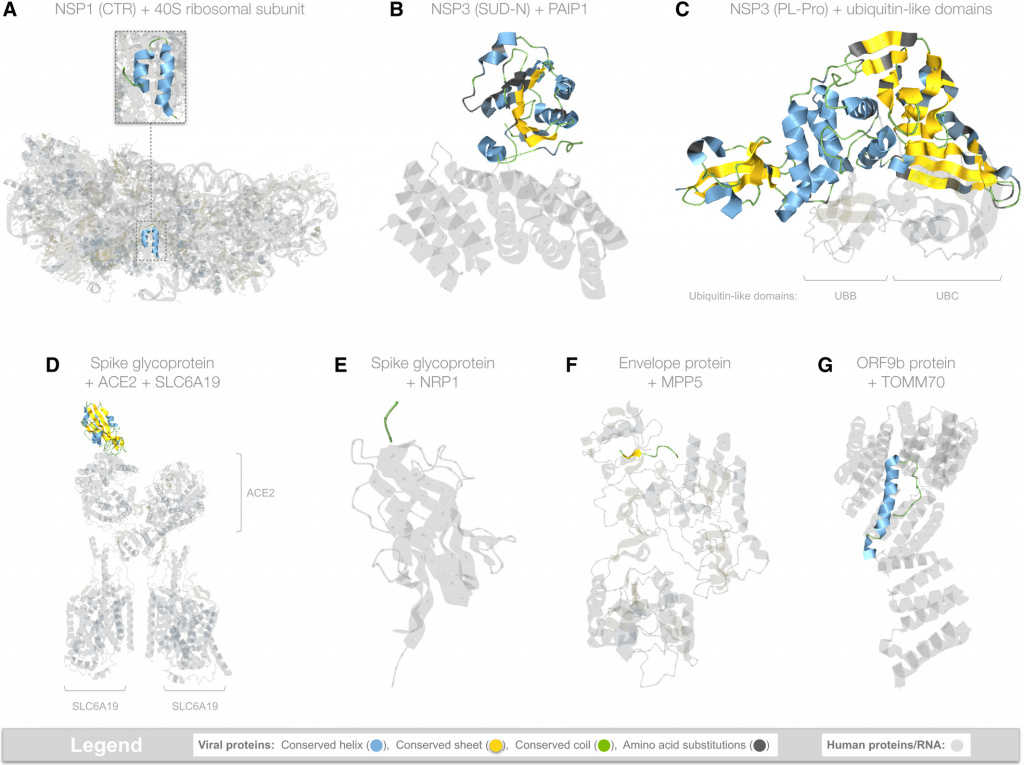 Credit: Seán I O’Donoghue, et al., 2021; DOI:
10.15252/msb.202010079 | CC BY 4.0
Credit: Seán I O’Donoghue, et al., 2021; DOI:
10.15252/msb.202010079 | CC BY 4.0
Третью группу составили 29% «командных» (teams) вирусных белков, потенциально способных к взаимодействию с другими вирусными белками. В одну команду попали все белки из репликационного и трансляционного комплексов, во вторую белки NSP10, NSP14 и NSP16.
Четвертая группа «подозреваемых» (suspects) — это оставшиеся 14 белков, для структур которых пока нет данных о взаимодействии с хозяйскими структурами или с другими вирусными белками. Семь белков из этой группы не имеют не только установленных экспериментально структур, но и возможных прототипов для моделирования. Эти белки отнесли к «темной области» (dark region) протеома.
Авторы отметили, что темная область в протеоме SARS-CoV-2 составила всего 31%, тогда как в среднем для вирусных протеомов в базе SwissProt (раздел UniProt c проверенными и аннотированными вручную геномами) такая область составляет 54%. Это говорит о том, что в настоящее время коронавирусы относительно хорошо изучены.
«Наш ресурс поможет исследователям понять, чем новые штаммы вируса отличаются друг от друга. Это кусочек головоломки, который, как мы надеемся, поможет справиться с новыми вариантами по мере их появления», — говорит руководитель группы Шон О’Донохью, профессор Института медицинских исследований в Гарване (Австралия).
Источник
Seán O’Donoghue, et al. SARS-CoV-2 structural coverage map reveals viral protein assembly, mimicry, and hijacking mechanisms // Molecular Systems Biology (2021)17:e10079; DOI: 10.15252/msb.202010079
Цитата по пресс-релизу

Вам будет интересно

 99
99
 0
0



 2034
0
2034
0
 1198
0
1198
0