Меню
Меню





 Все темы
Все темы
Создан органоид — модель мозга при синдроме ломкой X-хромосомы
Американские ученые создали органоид, моделирующий передний мозг пациента с синдромом ломкой X-хромосомы. На органоиде они показали, что при этом заболевании в переднем мозге нарушается нейрогенез, созревание нейронов, а также изменяется их возбудимость, и обнаружили новые потенциальные мишени лекарств.
Синдром ломкой X-хромосомы, или синдром Мартина–Белл, обусловлен утратой РНК-связывающего белка FMRP, который задействован в регуляции трансляции некоторых мРНК. Это заболевание является наиболее распространенной наследственной формой умственной отсталости и расстройств аутистического спектра.
Чаще всего в основе синдрома ломкой X-хромосомы лежит увеличение копийности повторов CGG в 5’-некодирующей области гена FMRP1, что приводит к гиперметилированию его промотора, прекращению экспрессии и отсутствию функционального белка FMRP в клетках. (Нарушение метилирования приводит к сужению соответствующего участка Х-хромосомы, видимой под микроскопом, что и дало синдрому название.)
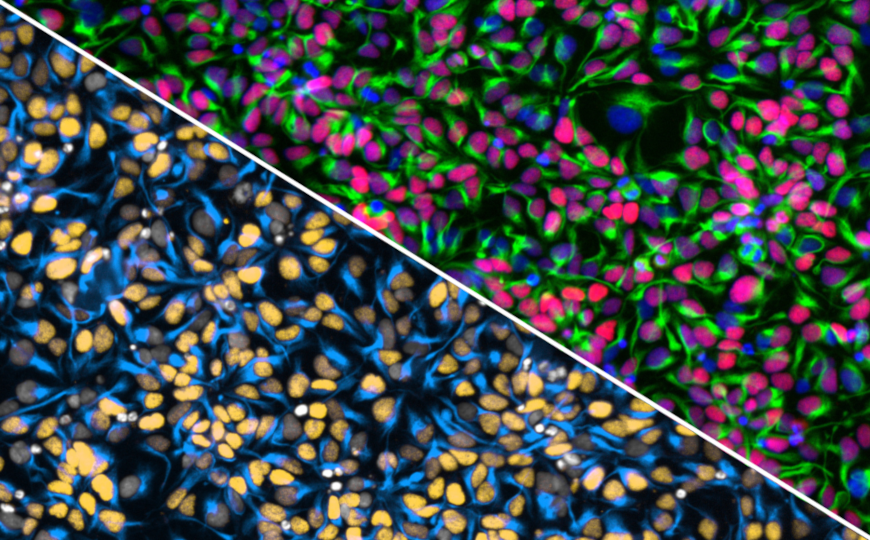
Чтобы исследовать нарушения, происходящие в переднем мозге пациентов с синдромом ломкой X-хромосомы, на клеточном и молекулярном уровне, американские ученые создали 3D-органоид, имитирующий передний мозг пациентов. Результаты исследования опубликованы в журнале Nature Neuroscience.
Для получения органоида использовали индуцированные плюрипотентные стволовые клетки, полученные из клеток человека с синдромом ломкой X-хромосомы. Авторы исследования отметили, что в органоидах наблюдается пониженный уровень пролиферации клеток-предшественников нейронов, дифференцировка нейронов нарушена, повышено количество синапсов, нейроны отличаются повышенной возбудимостью, при этом количество ГАМКергических нейронов в органоидах понижено по сравнению с нормой.
Детальное изучение клеток органоидов с помощью транскриптомного анализа, как на уровне всего органоида, так и на уровне отдельных клеток, выявило нарушения, происходящие при синдроме ломкой X-хромосомы на молекулярном уровне, в частности, множественные нарушения в экспрессии генов, связанных с развитием нервной системы.
Предыдущие исследования заболевания на модельных мышах показали, что устранить нарушения развития нервной ткани и формирования синапсов может фармакологическое ингибирование сигнального пути метаботропного рецептора глутамата 5 (mGluR5). Однако в настоящем исследовании, в котором был смоделирован именно мозг больного человека, а не мыши, этот результат подтвердить не удалось. Положительный эффект имело фармакологическое ингибирование сигнального пути фосфоинозитид-3-киназы (PI3K).
Наконец, чтобы определить, какие именно мРНК связывает белок FMRP, ученые выполнили ко-иммунопреципитацию FMRP со связанными мРНК из образцов нервной ткани, взятой от органоидов, соответствующих нормальному переднему мозгу человека, а также от эмбрионального мозга мыши, стадия развития которого примерно соответствовала органоиду, имитирующему мозг человека. Авторам удалось выявить ряд мРНК-мишеней FMRP, специфичных для человека. Одна из таких мРНК, кодирующая белок CHD2, вносит вклад в нарушение экспрессии генов в органоидах, имитирующих мозг пациента с синдромом ломкой X-хромосомы.
Ученые отмечают, что ряд мРНК-мишеней FMRP могут быть и мишенями фармакологического воздействия для устранения дефектов, ассоциированных с синдромом ломкой X-хромосомы.
Источник
Kang, Y., et al. A human forebrain organoid model of fragile X syndrome exhibits altered neurogenesis and highlights new treatment strategies // Nature Neuroscience, published 19 August 2021, DOI: 10.1038/s41593-021-00913-6.





 0
0