Меню
Меню





 Все темы
Все темы
Спорадическую болезнь Альцгеймера смоделировали на нейронах из фибробластов пациентов
Большинство моделей болезни Альцгеймера соответствуют ее наследственной форме, хотя гораздо более распространена спорадическая (ненаследственная) форма с поздним началом. Ученые из США разработали модель спорадической болезни Альцгеймера, перепрограммировав фибробласты пациентов с помощью микроРНК и транскрипционных факторов. В 3D-культурах полученных нейронов проявлялись все характерные признаки болезни. На этой модели авторы показали, что ингибиторы гамма- и бета-секретаз эффективны только до появления отложений амилоида бета. Кроме того, при спорадической форме болезни Альцгеймера пользу приносит подавление экспрессии ретротранспозонов.
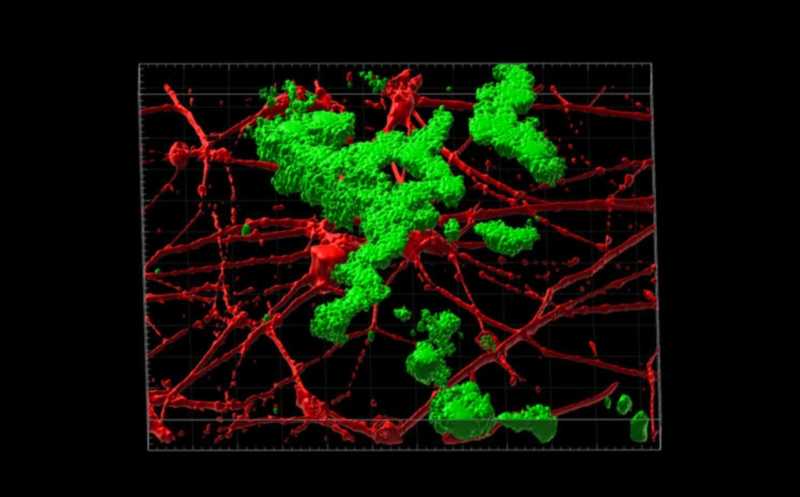
Трехмерная реконструкция отложения бляшек амилоида бета (зеленый) между нейронами, выращенными in vitro (красный). Эти нейроны были получины из фибробластов кожи пациентов со спорадической болезнью Альцгеймера.
Credit:
Andrew Yoo and Zhao Sun/Washington University in St. Louis | Пресс-релиз
Для изучения болезни Альцгеймера (БА) активно используют трансгенные клеточные и животные модели, но они на самом деле отражают только наследственную форму болезни с ранним началом. В реальности на нее приходится меньше 5% случаев, в то время как остальные случаи — это несемейная (спорадическая) болезнь Альцгеймера с поздним началом, риск которой повышается с возрастом. Ее трудно смоделировать из-за большого числа факторов, которые могут влиять на развитие болезни: старение, генетические факторы риска, менее значимые, чем те, что становятся причиной наследственных форм БА, и т.д.
Модели на основе индуцированных плюрипотентных клеток (иПСК) получают большое распространение, поскольку могут быть специфичными для каждого пациента. Если получить от пациента с БА соматические клетки (например, фибробласты), превратить их в иПСК и затем — в нейроны, то будут наблюдаться характерные для болезни проявления: отложения амилоида-бета, гиперфосфорилирование белка тау, окислительный стресс и др. Однако спорадическую болезнь Альцгеймера таким образом трудно смоделировать в том числе потому, что для развития некоторых характерных для нее признаков (появление нерастворимого тау, нейродегенерация) требуется много времени. Нейроны, полученные из иПСК, соответствуют фетальным нейронам, поэтому для моделирования поздней болезни Альцгеймера их нужно состарить. Команда под руководством исследователей из Медицинской школы Университета Вашингтона в Сент-Луисе решила эту проблему, создав модель спорадической формы болезни Альцгеймера в 3D культуре.
Ученые отобрали фибробласты от 10 пациентов с БА (4 случая с наследственной формой, 6 — со спорадической) и от 17 здоровых контролей. Затем фибробласты репрограммировали в нейроны коры головного мозга с помощью микроРНК 9/9* и 124, которые способствуют реконфигурации хроматина, «стиранию» программы фибробластов и активации нейрональной программы. Помимо этого, для получения нейронов применяли транскрипционные факторы NEUROD2 и MYT1L. В результате формировались электрически активные кортикальные нейроны. Их выращивали в 3D-культур двух типов: в плоском тонком слое геля (для анализа морфологии) и в сфероидах (для анализа поведения нейронов в условиях высокой плотности клеток).
Для начала исследователи смоделировали с помощью такого подхода наследственную форму болезни Альцгеймера (с мутациями в генах PSEN1 или APP). В культурах таких нейронов содержалось больше, чем в контроле, внеклеточных отложений амилоида-бета, причем в сфероидах их накапливалось больше, чем в плоском геле. Отложение амилоида-бета уменьшалось при действии ингибиторов бета- и гамма-секретаз, которые участвуют в разрезании его белка-предшественника APP.
В 3D-моделях наследственной формы БА также наблюдалось гиперфосфорилирование белка тау — накопление его изоформ 3R и 4R, которые выходят в состав нейрофибриллярных клубков. И в плоском геле, и в сфероидах появлялась спонтанная нейродегенерация; сфероиды с патогенными мутациями при культивировании уменьшались в размерах сильнее, чем контроль. И таупатия, и нейродегенерация зависели от появления отложений амилоида-бета, так как ингибиторы бета- и гамма-секретаз значительно уменьшали их проявления.
Модели наследственной формы БА также имели особые характеристики на транскриптомном уровне. В них была повышена экспрессия генов MMP3, IBSP, TAGLN, ассоциированных с повышенным риском развития БА, а также генов, связанных с нейровоспалением, — CCL20, CSF3, IL10. Таким образом, адекватность моделей БА подтвердилась.
На следующем этапе исследователи репрограммировали в кортикальные нейроны фибробласты, полученные от пациентов с поздней болезнью Альцгеймера. В отличие от большинства существующих моделей этой формы болезни, в этой модели проявлялись все ее характерные признаки: отложения амилоида-бета, таупатия, нейродегенерация. При этом на количество внеклеточных отложений амилоида бета значительно влиял возраст пациента: так, в нейронах из фибробластов 22-летнего пациента они были минимальными, в нейронах от более старых пациентов были выражены сильнее. Сфероиды от пожилых здоровых доноров показали некоторое отложение амилоида, но гораздо меньшее, чем сфероиды от пациентов.
В модели поздней БА тоже присутствовали изоформы 3R и 4R гиперфосфорилированного тау белка и его агрегаты, склонные провоцировать агрегацию других молекул того же белка. И в плоском геле, и в сфероидах происходила спонтанная нейродегенерация. Так, за 16 дней культивирования сфероиды, моделирующие БА, уменьшались в размерах на 50%, а контрольные сфероиды — только на 10–20%. Исследователи также зафиксировали дисфункцию образования синапсов в их модели.
Необычными оказались результаты ингибирования образования амилоида бета. Если клетки обрабатывали ингибиторами бета- или гамма-секретазы на достаточно ранних сроках культивирования (16 дней), то агрегаты амилоида-бета уменьшались до 90%. Однако при более поздней обработке (например, на 22-й день, когда отложения амилоида бета уже образовались) эффективность ингибиторов исчезала практически полностью. Это говорит о том, что для предотвращения развития БА нужно начинать действовать до образования отложений амилоида бета.
Транскриптомный анализ моделей спорадической формы БА показал повышение экспрессии ряда генов, так или иначе связанных с воспалением и иммунным ответом. Например, была повышена экспрессия генов матриксных металлопротеаз (MMP1, MMP3, MMP8). При этом нокдаун этих генов, по отдельности или всех вместе, никак не влиял на отложения амилоида-бета, фосфорилирование тау или нейродегенерацию. Между моделями наследственной и спорадической форм болезни Альцгеймера нашлись и сходства: в частности, в них одинаково нарушалась экспрессия генов, ассоциированных с регуляцией образования синапсов, аутофагией, выработкой цитокинов и т. д.
В репрограммированных нейронах была нарушена экспресия ретротранспозонов, причем нарушения становились более явными при старении. Изменения в экспрессии ретротранспозонов могут приводить к нейровоспалению, накоплению аномального белка тау и ассоциированной нейротоксичности. Ученые попытались воздействовать на ретротранспозоны с помощью ламивудина — ингибитора обратной транскриптазы. Это не только снизило нейродегенерацию в сфероидах, но и уменьшило образование агрегатов амилоида бета и белка тау. Повреждение ДНК, вызванное активностью ретротранспозонов, также снизилось, а транскриптомный анализ указал на уменьшение воспаления.
Что интересно, ламивудин оказался полезен в модели спорадической формы БА, но когда им обрабатывали нейроны и сфероиды с наследственной формой болезни, это практически никак не влияло на ее проявления. Следовательно, аномалии, связанные с ретротранспозонами, более значимы именно в спорадической форме болезни.
Олигодендроциты производят амилоид бета при болезни Альцгеймера
Источник
Sun, Z. et al. Modeling late-onset Alzheimer’s disease neuropathology via direct neuronal reprogramming. // Science, 385 (2024). DOI: 10.1126/science.adl2992

Вам будет интересно

 93
93
 0
0


 92
0
92
0
 112
0
112
0
 132
0
132
0