Меню
Меню





 Все темы
Все темы
Вирусы эукариот и бактериофаги защищаются от иммунитета хозяина одинаковыми способами
Быстрая эволюция вирусов сформировала широкое разнообразие их белковых последовательностей, многие из которых пока слабо охарактеризованы — это затрудняет как изучение вирусов, так и борьбу с ними. Исследовательская группа, возглавляемая Дженнифер Дудной, предсказала структуры более чем 67 тысяч вирусных белков. Оказалось, что почти две трети из них не имели гомологов в базе данных AlphaFold, однако у многих оставшихся обнаружилось выраженное структурное сходство с белками организма-хозяина, которое, по-видимому, помогает вирусам инфицировать клетки. Кроме того, ученые описали механизм уклонения от иммунной реакции хозяина и показали, что он характерен как для вирусов эукариот, так и для бактериофагов.
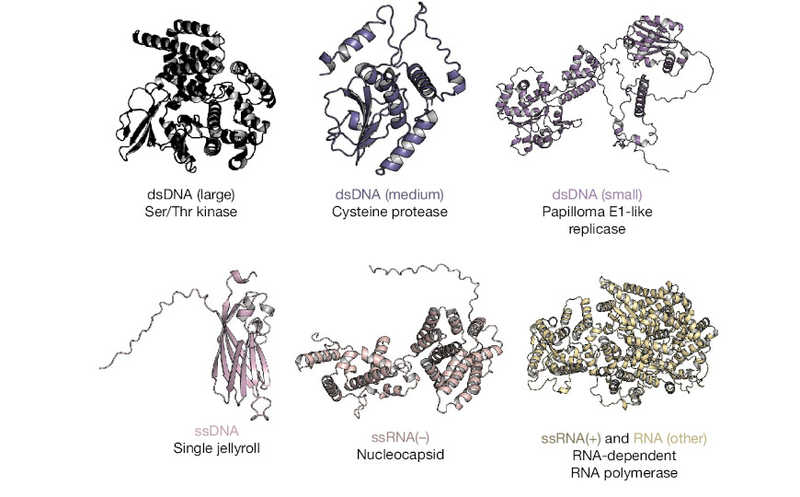
Структуры белков, принадлежащих к самому таксономически разнообразному кластеру.
Credit:
Nature (2024). DOI:
10.1038/s41586-024-07809-y |
CC BY
Вирусные белки необходимы им для инфицирования и репликации. Некоторые белки или их домены высококонсервативны и широко распространены в разных семействах вирусов, однако из-за быстрой эволюции общее разнообразие последовательностей очень высоко. Это затрудняет понимание механизмов вирусной инфекции и эволюции, поскольку функции многих белков до сих пор не известны. Коллектив под руководством нобелевского лауреата Дженнифер Дудны задался целью устранить этот пробел — исследователи предсказали структуры почти 70 тысяч вирусных белков и обнаружили, что 62% этих структур не имеют гомологов в базе данных AlphaFold. Результаты работы опубликованы в журнале Nature.
Ученые включили в собранную ими базу данных 67 715 белков, встречающихся у 4 436 видов вирусов эукариот. Для предсказания их структур, основанного на множественном выравнивании последовательностей, они воспользовались ColabFold. Эти белки кластеризовали по последовательности, а затем по структуре, что позволило получить 18 192 белковых кластеров, 12 422 из которых содержали только по одному белку.
Чтобы охарактеризовать таксономическое распределение представителей полученных кластеров, авторы работы провели их структурное выравнивание с 2,3 миллионами белков из базы данных AlphaFold. Оказалось, что 62% вирусных белков не имели в ней гомологов и встречались только в одном семействе вирусов. Это свидетельствует о том, что эволюция вирусов порождает значительное число новых белковых структур, отсутствующих в современных базах данных. При этом многие представители выделенных белковых кластеров обладали похожей структурой, несмотря на низкое сходство последовательностей.
Исследователи решили подробнее проанализировать ДНК-связывающие белки, которые находят применение в биотехнологии, в диагностике и редактировании генома. Сначала они изучили TATA-связывающие белки (TBP), которые распознают мотивы TATA-box в эукариотических промоторах. Многие ДНК-вирусы используют TBP, чтобы стимулировать экспрессию собственных генов или модулировать экспрессию генов хозяина. К настоящему моменту известны три семейства вирусов с двуцепочечным ДНК-геномом, кодирующих такие белки. Теперь же ученые нашли свидетельства наличия таких белков еще в четырех семействах ДНК-вирусов. Они также исследовали семейство I3L — белки поксвирусов, связывающие одноцепочечную ДНК. Предполагается, что I3L участвуют в репликации или репарации вирусной ДНК, однако структура этого белка еще не была получена экспериментально, а его связь с различными белковыми укладками и семействами остается неизвестной. Дудна и соавторы обнаружили в белке I3L олигонуклеотид-связывающую укладку, аналогичную таковой у ДНК-связывающих белков бакуловируса или фага T7. Аналогичную укладку они выявили еще в четырех семействах ДНК-вирусов.
Структурные особенности часто оказываются куда более консервативны, чем последовательности нуклеотидов или аминокислот. Ученые проверили, может ли структурное выравнивание белков человеческих вирусов дать представления о функциях плохо аннотированных белков, кодируемых их геномом. Выравнивание созданной ими базы данных с первоначальной версией AlphaFold, содержащей более 300 тысяч белков из 21 организма (эукариот, бактерий и архей) выявило повсеместное структурное сходство, которое было распространено даже при низкой гомологии последовательностей. Из предсказанных вирусных белков 14 531 был выровнен на один из членов базы данных AlphaFold, причем преимущественно на эукариотические белки. Например, ряд белков поксвирусов имел структурное сходство с аутоингибиторным доменом гасдерминов — белков, вовлеченных в пироптоз. Еще несколько поксвирусных белков были схожи с галактозилтрансферазой человека, которая, как считается, обеспечивает связывание вируса с поверхностными гликозаминогликанами для проникновения в клетку. Эти и другие обнаружения указывают на высокое сходство структуры многих вирусных и невирусных белков, причем такие белки кодируются вирусами с разным типом генома. Эволюционное происхождение такой структурной консервативности не вполне ясно, однако авторы исследования предполагают, что многие кластеры, содержащие белки разнообразных по строению генома вирусов, возникли в результате горизонтального переноса генов.
Многие аспекты иммунитета эукариот и прокариот имеют общее происхождение. Примером служат пути cGAS-STING у многоклеточных животных и антифаговые сигнальные системы циклических олигонуклеотидов (CBASS) у прокариот. В обоих случаях белковый сенсор детектирует вирусный сигнал и генерирует нуклеотидный вторичный мессенджер, активирующий противовирусный ответ. У вирусов при этом сформировались свои способы уйти от хозяйского иммунитета — так, у фага T4 имеется анти-CBASS белок 1 (Acb1), который разрушает сигнальные циклические олигонуклеотиды в зараженной клетке. Этот белок относится к РНК-лигаза-Т-подобным фосфодиэстеразам (Lig-T-подобным PDE). Некоторые РНК-вирусы эукариот кодируют PDE, способные разрушать 2′,5′-олигоаденилаты, причем их укладка аналогична таковой у Acb1. Исследователи задались вопросам, не являются ли такие белки распространенной системой, с помощью которой вирусы уклоняются от иммунной защиты. Структурный и филогенетический анализ показал, что у вирусов эукариот имеется несколько независимых ветвей Lig-T подобных PDE. Среди них авторы обнаружили подргуппу, которая хорошо экспрессируется в клетках млекопитающих. На основе клеток HEK293T они получили систему, в которой STING можно активировать обработкой 2′,3′-cGAMP или ненуклеотидным агонистом diABZI, а саму активацию отследить по экспрессии люциферазы. Оказалось, что LigT-подобные PDE птичьих поксвирусов подавляли сигналы 2′,3′-cGAMP и препятствовали активации STING, но слабо влияли на активацию с помощью diABZI. Это подавление требовало ферментативной активности PDE, поскольку мутация их каталитических гистидинов существенно ослабляла такой эффект. Дальнейший анализ показал, что LigT одного из представителей — вируса оспы голубей — расщепляет различные варианты cGAMP аналогично тому, как это делает Acb1 фага T4. Иными словами, опираясь на структурное сходство, Дженнифер Дудна и соавторы открыли новый механизм деградации 2′,3′-cGAMP вирусами эукариот. Они пришли к выводу, что его расщепление с помощью LigT-подобных PDE — один из общих вирусных механизмов защиты от иммунитета хозяина.
Созданная в ходе данной работы база данных структур вирусных белков открывает возможности для выявления механизмов взаимодействия «вирус—хозяин», что позволит углубить понимание эволюции вирусов и может лечь в основу разработки новых мер борьбы с вирусными инфекциями.
Источник
Nomburg, J., et al. Birth of protein folds and functions in the virome. // Nature (2024). DOI: 10.1038/s41586-024-07809-y





 0
0