
 Меню
Меню





 Все темы
Все темы
Внутриутробная терапия смягчила симптомы болезни Помпе
Терапию при болезни Помпе — наследственной лизосомной болезни накопления — нужно начинать как можно раньше, но при самых неблагоприятных обстоятельствах повреждения сердца становятся необратимыми уже при рождении. Исследователи из Калифорнийского университета в Сан-Франциско и коллабораторы разработали протокол для начала ферментозаместительной терапии in utero и успешно воплотили его в жизнь. На момент написания новости пациентке уже 16 месяцев и она развивается нормально.

Болезнь Помпе — это наследственная лизосомная болезнь накопления. Больные дети обычно рождаются с увеличенным сердцем и без лечения не живут дольше двух лет. Заболевание редкое, менее одного случая на 100 000 рождений. Причина — мутация в гене, кодирующем кислую альфа-глюкозидазу, фермент, который расщепляет гликоген. Без фермента гликоген накапливается в мышцах и клетках сердца. Очень важно как можно раньше начать лечение, но иногда повреждения сердца становятся необратимыми уже на ранних этапах развития, то есть еще до рождения.
Исследователи из Калифорнийского университета в Сан-Франциско и коллабораторы разработали протокол, который позволяет ввести рабочую копию фермента пациенту in utero. Такую терапию на человеке проводили впервые.
Ферментозаместительную терапию (ФЗТ) in utero исследователи изначально опробовали на мышах, после чего получили разрешение на начало фазы 1 клинических испытаний ФЗТ in utero для восьми лизосомных болезней накопления, которые проявляются еще до родов.
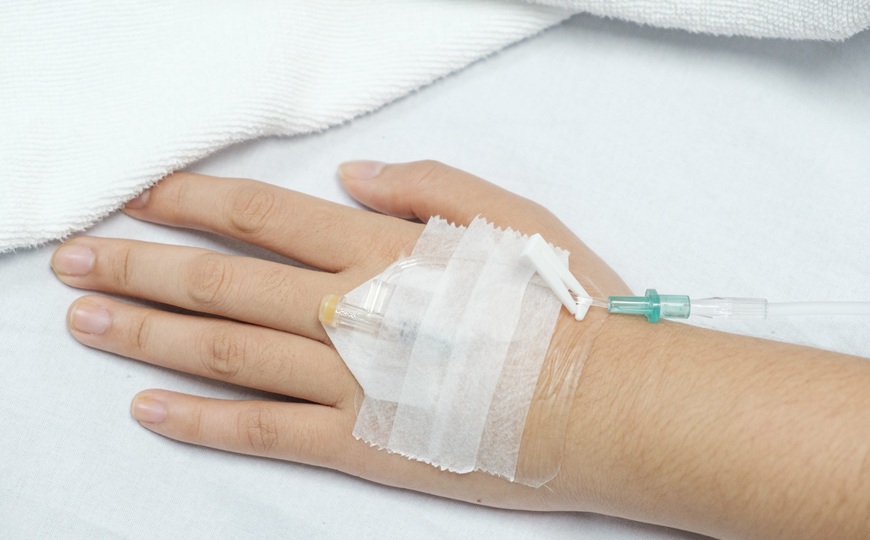
Алглюкозидазу альфа (20 мг на кг предполагаемого веса плода) вводили в пупочную вену, начиная с 24 недель беременности. Всего препарат вводили шесть раз с перерывами в две недели. Каждый раз отбирался небольшой образец крови для того, чтобы подтвердить попадание в пупочную вену и проконтролировать ход терапии. После рождения ребенок получал стандартную терапию для болезни Помпе. У матери и девочки не было побочных реакций на введение препарата.
Креатинкиназа может быть маркером долговременного повреждения мышц. У пациентки креатинкиназа была в норме после рождения, в отличие от других детей с таким же состоянием, у которых этот параметр был повышен до начала терапии. У девочки не было признаков двигательных дефектов, ребенок развивался нормально и вовремя достигал всех этапов развития.
Эхокардиограммы пациентки также были нормальными, как и размеры сердца, что выделялось на фоне результатов обследования детей с болезнью того же типа, что и у девочки. При болезни Помпе, проявляющейся во младенчестве, отложения гликогена видны уже в плаценте. После ФЗТ in utero свидетельств отложений не наблюдалось. Антитела к препарату обнаруживались уже после третьего введения in utero, однако они оставались на уровне, ниже клинически значимого.
Таким образом, авторы показали безопасность протокола ФЗТ in utero как для матери, так и для плода. На момент написания статьи девочке было 13 месяцев, а выхода новости — 16 месяцев, и ее развитие идет нормально. Врачам удалось замедлить или даже остановить прогрессирование заболевания во время внутриутробного развития. По мнению авторов, введение фермента in utero также может натренировать иммунную систему ребенка так, чтобы она не вырабатывала антитела на искусственный фермент, как иногда происходит у детей, получающих ФЗТ.
Источник:
Jennifer L. Cohen, et al. In Utero Enzyme-Replacement Therapy for Infantile-Onset Pompe’s Disease // NEJM (2022), published November 09, 2022, DOI: 10.1056/NEJMoa2200587

Вам будет интересно
 146
146
 0
0


 292
0
292
0
 673
0
673
0








