Меню
Меню





 Все темы
Все темы
Белки непохожих вирусов обладают похожей 3D-структурой
Авторы статьи в Science Advances представили базу данных Viral AlphaFold Database, которая содержит десятки тысяч структур вирусных белков, предсказанных с помощью AlphaFold 2. Оказалось, что многие пространственные структуры консервативны даже среди вирусов, заражающих разные организмы — бактерии, археи, различные виды эукариот. Ученые также описали еще не аннотированные белки, не имеющие структурных аналогов, и смогли предположить функции некоторых из них. Viral AlphaFold Database — ценный ресурс для изучения эволюции вирусов, предсказания функций неизвестных белков и разработки стратегий борьбы с вирусами.
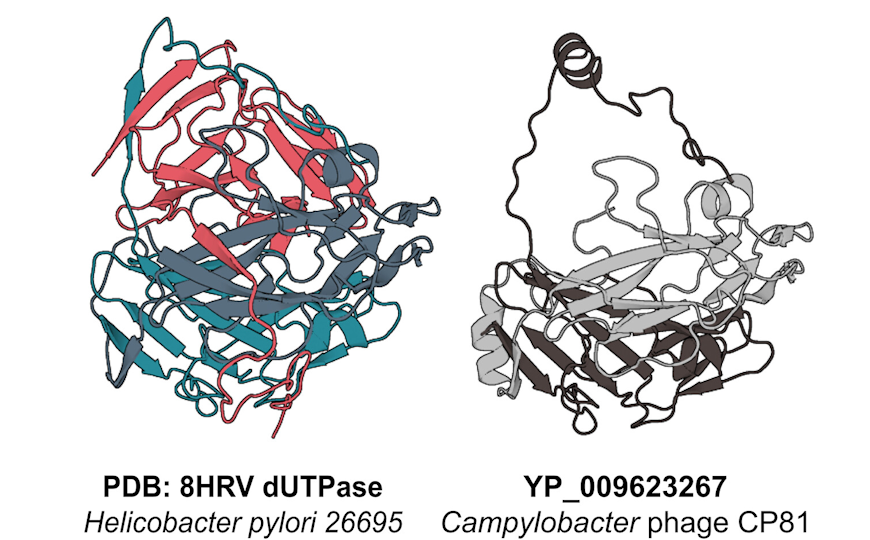
Сходство олигомеризации может наблюдаться даже у белков, аминокислотные последовательности которых сильно различаются.
Credit:
Science Advances (2025). DOI:
10.1126/sciadv.adz8560 |
CC BY
Вирусы — наиболее генетически разнообразная биологическая группа, однако подавляющее большинство их белков остается функционально неохарактеризованными. Экспериментальное определение трехмерных структур таких белков осложнено особенностями их трансляции: зачастую с вирусного генома одним транскриптом считывается конкатенированная цепь белков — полипротеин, который впоследствии разрезается на отдельные белки. Поэтому в существующих ресурсах предсказанные модели вирусных белков представлены крайне ограничено. Чтобы восполнить этот пробел, авторы статьи в Science Advances собрали набор белковых последовательностей, принадлежащих вирусам бактерий, архей и эукариот, при помощи AlphaFold 2 построили для них трехмерные модели, включая как одиночные белки, так и возможные гомодимеры.
Используя последовательности из базы RefSeq, ученые создали ресурс Viral AlphaFold Database (VAD), включающий около 27 тысяч белковых структур и столько же моделей гомодимеров. Предсказание структуры проводили с помощью AlphaFold 2, и стандартный алгоритм этой модели моделировал пространственную укладку белков с высокой степенью достоверности в большинстве случаев. Сравнение с другими базами данных вирусных белков подтвердило, что VAD отличается более высоким качеством моделей.
Следующим шагом стало выявление сходных структурных мотивов. Авторы кластеризовали белки по сходству их трехмерной структуры. Это позволило обнаружить множество консервативных фолдов, схожих у вирусов бактерий, архей и эукариот. Так, 25,5% белков бактериофагов имели структурные аналоги в других организмах, для вирусов эукариот этот показатель составил 31,1%, а для вирусов архей — 37,6% В ряде случаев эти сходства были не заметны на уровне последовательности, что подчеркивает ценность структурного подхода.
Особое внимание авторы уделили предсказанию гомодимерных структур, поскольку взаимодействия между одинаковыми белковыми субъединицами зачастую играют ключевую роль в функционировании белка. Используя AlphaFold-Multimer, исследователи смоделировали димерные взаимодействия почти всех вирусных белков из базы (кроме 200 самых крупных) и сравнили их с известными структурами в Protein Data Bank. Структуры многих предсказанных гомодимеров согласовались с экспериментально определенными. Особенно консервативными гомодимерами оказались пептидил-тРНК гидролазы (Pth) — они имеются у бактериофагов, а также у вирусов птиц и насекомых. Эти ферменты расщепляют пептиды, связанные с тРНК, что предотвращает остановку рибосом и освобождает тРНК для трансляции.
Исследователи выявили ранее не аннотированные белки, не имеющие структурных аналогов в существующих базах данных. Некоторые из них удалось связать с потенциальными биологическими функциями. В частности, ученые идентифицировали профаг, который кодирует систему токсин-антитоксин II типа в клетках бактерий. Такие системы обеспечивают отбор дочерних клеток бактерий, унаследовавших определенную плазмиду, и ранее эта система у вирусов описана не была.
Другим интересным открытием стало обнаружение консервативных фолдов в фаговых системах защиты и контрзащиты. Эти системы используются бактериофагами и некоторыми вирусами эукариот для борьбы с вирусами-конкурентами. Анализируя VAD, авторы выявили структуры, схожие с антифаговыми белками (например, DarG и gp28) и с анти-CRISPR-белками (AcrIIA8, AcrIA1). Эти структурные мотивы встречаются у вирусов с разными хозяевами и сохраняются даже при отсутствии сходства последовательностей, что указывает на их важную роль в эволюции иммунных систем. Таким образом, созданная база VAD может служить источником гипотез о функциях неизвестных белков и способствовать открытию новых механизмов в биологии вирусов.
Созданная база данных Viral AlphaFold Database представляет собой мощный инструмент для изучения вирусных белков. Она не только дополняет существующие ресурсы, но и открывает новые возможности для функциональной аннотации, поиска эволюционных связей и предсказания белковых взаимодействий. Работа показывает, что вирусы, несмотря на колоссальное разнообразие, обладают консервативными структурными мотивами, и знания об этих универсальных элементах важны для разработки противовирусной терапии и для понимания эволюции вирусов.
Вирусы эукариот и бактериофаги защищаются от иммунитета хозяина одинаковыми способами



 0
0