Меню
Меню





 Все темы
Все темы
Число повторов в гене гентингтина растет не только в нейронах, но и в клетках крови
Болезнь Гентингтона — тяжелое нейродегенеративное заболевание, при котором возраст проявления симптомов зависит от унаследованного пациентом от родителя количества повторов CAG в гене гентингтина (HTT). Но кроме того, в нейронах пациента происходит соматическая экспансия повторов, которая и приводит к экспрессии токсичной формы белка и нейродегенерации. Ученые из Великобритании, США и Швеции установили, что число повторов в гене HTT растет и в клетках крови, и это можно использовать для ранней диагностики прогресссирования болезни, задолго до проявления функциональных нарушений.

Международная группа под руководством ученых из Университетского колледжа Лондона показала, что первые изменения в мозге, вызванные болезнью Гентингтона, появляются за десятки лет до появления клинических симптомов. Кроме того, экспансия повторов в гене гентингтина HTT, выявленная в клетках крови, может свидетельствовать о начале быстрого прогрессирования болезни.
Болезнь Гентингтона (HD) — аутосомно-доминантное нейродегенеративное заболевание, при котором летальный исход наступает в среднем через 10–13 лет после проявления симптомов. Оно ведет к потере средних шипиковых нейронов и нейродегенерации полосатого тела — структуры мозга, регулирующей мышечный тонус и участвующего в формировании условных рефлексов. В настоящее время не существует методов лечения, изменяющих течение этого заболевания.
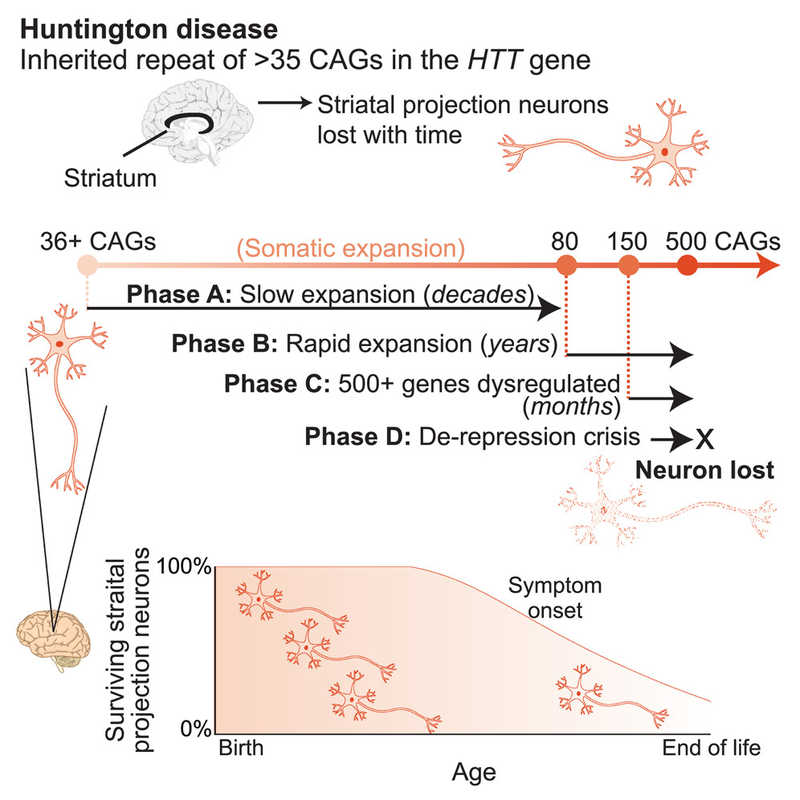
Причиной болезни является экспансия (увеличение количества) повторов CAG в гене гентингтина (HTT). При этом в мутантном белке увеличивается количество глутаминовых остатков (образуется полиглутаминовый участок, или полиQ); такая форма белка токсична для нейронов. Подобные мутации называют динамическими из-за продолжительного роста числа повторов в ряде поколений. Они возникают из-за ошибок репликации; увеличение количества повторов вызвано проскальзыванием (смещением нитей ДНК друг относительно друга) или образованием аномальных форм ДНК, нарущающих репликацию. Для болезни Гентингтона, как и для некоторых других наследственных неврологических заболеваний, известно явление антиципации — усиление симптомов и (или) более раннее проявление у детей по сравнению с родителями; с каждом поколением болезнь становится тяжелее. Причиной являются именно накопление повторов. Патологическим считается количество CAG-повторов, превышающее 36 (27–35 повторов могут говорить о риске для последующих поколений).
У пациентов с болезнью Гентингтона нейроны полосатого тела наиболее уязвимы; в них происходит соматическая экспансия повторов. Авторы исследования, которое недавно было опубликовано в Cell, обнаружили, что число повторов в гене нейрона за десятилетия его жизни может увеличиться на порядок (с 40–45 до 100–500+), причем белок становится токсичным при 150 CAG и более — мутации с меньшим количеством повторов остаются «биологически тихими». Динамика этого процесса соответствует долгому латентному периоду и быстрому прогрессированию болезни. Это явление приводит к соматическому мозаицизму (различию числа повторов в разных типах клеток у одного пациента). В клетках, которые, подобно нейронам, не делятся, изменения длины повторов могут возникнуть при случайном смещении нитей во время транскрипции или дестабилизации двойной спирали, с последующим неправильным спариванием и репарацией, восстанавливающей комплементарность. Почему соматическая нестабильность гена гентингтина особенно сильно выражена в нейронах определенного типа, пока неясно.
 Credit: Cell.2025. DOI:
Credit: Cell.2025. DOI:
Таким образом, соматическая экспансия повторов CAG оказывает ключевое влияние на прогрессирование болезни Гентингтона. Как моногенное заболевание, она представляется перспективной мишенью для терапии. И мутации в соматических клетках, и белки активно изучаются в качестве терапевтических целей. (Подробнее о кандидатных препаратах, действующих на мРНК гентингтина.) Но необходимо развивать диагностику, чтобы правильно выбрать наиболее подходящий момент для начала лечения. Двигательные симптомы становятся заметными, когда нейродегенерация в полосатом теле уже происходит. В то же время «тихие» мутации возникают раньше, чем появляется симптоматика. Однако предыдущие исследования связи между экспансией повторов CAG и ранним патологическим проявлением заболевания опирались на посмертный анализ мозга. Для ранней диагностики необходимо выявлять соматическую нестабильность CAG или другие биомаркеры в иных типах образцов.
Пациентов с экспансией гена болезни Гентингтона (HDGE) классифицируют согласно интегрированной системе стадирования HD (HD-ISS): стадия 0 — объем полосатого тела в пределах нормы, 1 — детектируется изменение объема полосатого тела и (или) его отделов, 2 — наличие двигательных и/или когнитивных признаков, 3 — начало функциональных нарушений. На стадиях 0 и 1 профилактическое лечение могло бы принести наибольшую выгоду.
Чтобы разработать методы обнаружения первых признаков нейродегенерации, вызванной соматической экспансией повторов в нейронах, до клинических проявлений, авторы нового исследования провели анализ когнитивных функций, структуры мозга, генетических маркеров и белков спинномозговой жидкости в уникальной когорте HD Young Adult Study (HD-YAS). В нее входят молодые люди, у которых прогнозировалось появление функциональных нарушений примерно через 23 года от начала исследования. В статье представлены результаты 4,5 лет наблюдения этой когорты.
Изначально в исследовании участвовал 131 человек (64 HDGE и 67 здоровых контролей), и 103 (57 HDGE и 46 контрольных) из них вернулись через 4,5 года для повторного наблюдения. Вместо тех, кто не вернулся, набрали еще 9 лиц с HDGE и 14 контролей; таким образом, всего в исследовании приняло участие 154 человека. В начале наблюдений из 54 человек с HDGE, для которых имелись данные нейровизуализации, 44 находились на стадии 0, девять человек — на стадии 1 и всего один — на стадии 2. Через 4,5 года 10 человек перешло со стадии 0 на стадию 1, прогрессирования до стадии 2 не наблюдалось.
Результаты нейровизуализации выявили более высокие показатели атрофии в скорлупе и хвостатом ядре полосатого тела у группы HDGE по сравнению с контролем. Признаки нейродегенерации коррелировали с экспансией CAG и возрастом. На стадиях 0 и 1 уже имелись признаки атрофии мозга, сопровождаемой незначительными микроструктурными изменениями в белом веществе.
Анализ 216 образцов спинномозговой жидкости (СМЖ) и плазмы крови, собранных в разных временных точках, показал, что в СМЖ группы HDGE по сравнению с контролем быстрее растет уровень белка Nfl (биомаркер повреждения нейронов) и снижается уровень белка PENK (биомаркер состояния MSN полосатого тела). Средние уровни Nfl во всех временных точках были повышены у участников, у которых болезнь прогрессировала из стадии 0 в стадию 1; после корректировки по возрасту и полу разница с участниками без прогрессии была статистически значимой.
Авторы проверили, коррелируют ли эти изменения с экспансией повторов CAG в клетках крови. По данным секвенирования образцов крови рассчитали коэффициенты соматической экспансии (SER), и они значительно увеличивались с течением времени во всех группах HDGE, включая стадию 0. При большей длине CAG-последовательности быстрее возрастал SER. Увеличение SER было ассоциировано с атрофией хвостатого ядра и скорлупы полосатого тела, а также с изменениями уровней NfL и PENK. Это говорит о том, что SER, измеренный для клеток крови, может быть косвенным количественным индикатором соматических мутаций в нейронах.
Результаты секвенирования также показали, что у некоторых участников были потеряны последовательности CCGCCA и CAACAG, которые в норме отделяют в гене HTT повторы CAG от повторов CCG, кодирующих полипролин. Участники с такой потерей демонстрировали более высокие показатели атрофии хвостатого ядра и скорлупы, а также более высокие уровни NfL и более низкие уровни PENK, что предполагает влияние этого участка ДНК на скорость невропатологических изменений.
Данная работа определяет надежные биомаркеры прогрессирования болезни Гентингтона и показывает, что экспансия повторов в ДНК клеток крови, скорее всего, соответствует специфической для конкретного пациента экспансии повторов в мозге. Она также подтверждает роль увеличения числа повторов в патогенезе болезни Гентингтона и их потенциальную роль как мишени для терапии. Идентификация пациентов, находящихся на начальных этапах прогрессирования болезни, поможет в организации клинических испытаний препаратов, модулирующих экспансию повторов и способных отсрочить или предотвратить клинически диагностируемые двигательные нарушения.
Аномалии в сосудах мозга позволяют обнаружить болезнь Гентингтона еще до развития симптомов
Источник
Scahill R., et al. Somatic CAG repeat expansion in blood associates with biomarkers of neurodegeneration in Huntington’s disease decades before clinical motor diagnosis // Nature Medicine (2025), published online 17 January 2025. DOI: 10.1038/s41591-024-03424-6
Вам будет интересно

 7
7
 0
0


 32
0
32
0