Меню
Меню





 Все темы
Все темы
Диагностика редких генетических болезней меняет стратегию лечения пациентов
Редкие генетические заболевания сложно диагностировать, и многие пациенты с такими болезнями много лет не получают необходимого лечения. Авторы статьи в журнале Genetics in Medicine Open продемонстрировали, как диагностика тяжелых редких генетических заболеваний в рамках проекта Deciphering Developmental Disorders (DDD) влияет на жизнь пациентов и его семей.
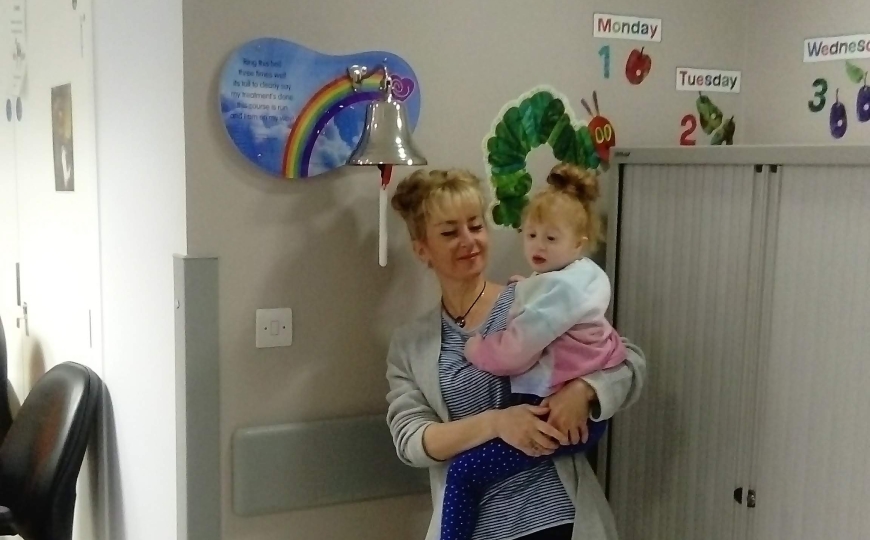
Участники DDD — Джейди и ее мать Лиза. Заболевание Джейди приводит к снижению иммунитета, ей рекомендованы пересадка костного мозга и дополнительные прививки.
Credit:
Несмотря на массовое внедрение генетического тестирования, диагностика редких генетических заболеваний по-прежнему остается проблемой. Группа ученых из Великобритании выясняла, как точная диагностика нарушений развития у детей влияет на жизнь пациентов и их семей.
Deciphering Developmental Disorders (DDD) — совместный проект Института Сэнгера и подразделений клинической генетики Национальной службы здравоохранения Великобритании (NHS). В проекте участвуют дети с тяжелыми генетическими нарушениями развития, которые ранее не удавалось диагностировать. Анализировали геномы трио — ребенка, матери и отца. Для диагностики использовали микрочипы CGH (хромосомный микроматричный анализ, ХМА) выявляющие вариации числа копий (CNV), потери хромосом или их протяженных участков, микрочипы для SNP-генотипирования, а также полноэкзомное секвенирование. С момента запуска проект уже привел к открытию 60 новых генетических нарушений, сообщается в пресс-релизе. В исследовании приняло участие более 13 500 семей, и в начале 2023 года диагноз был поставлен примерно 41 %.
Новая статья представляет данные исследования 4237 пациентов (среди них 47 % женского пола), которым смогли поставить диагноз, и их семей. Медианное время от набора участников до получения результата составило 3,4 года (диапазон 1,1–9,4 года), и возраст пациентов к моменту диагноза составлял от 1,8 до 55 лет (медиана 11 лет).
Данные об участниках и их родителях собрали с марта 2021 года по июль 2022 года. Патологии были описаны в терминах Human Phenotype Ontology. Также проводилось анкетирование, включающее вопросы о лечении, тестировании или скрининге, репродуктивном выборе родителей после получения диагноза и т.п.
В перечень диагнозов участников проекта вошли более 800 редких состояний, связанных как с моногенными мутациями, так и с мультигенными структурными вариантами. Наблюдались различные типы наследования: аутосомно-доминантное (68 % — мутации de novo, 7 % унаследованы от больного родителя и 8 % неуточненного происхождения), аутосомно-рецессивное (11 %), сцепленное с Х-хромосомой наследования (5 %), а также множественные диагнозы с различными типами наследования (1 %).
Клиническое ведение пациента после постановки диагноза в ходе DDD изменилось в 28 % случаев (у 1183 человек), то есть пациент начал получать другое лечение (143 человека), был направлен на дополнительное тестирование или скрининг. Пренатальное тестирование у взрослых пациентов, планирующих ребенка, в этот список не включалось, так как оно не относится напрямую к управлению здоровьем самого пациента.
Под изменением лечения подразумевалось как начало новой терапии, так и пересмотр уже существующей или отказ от определенных методов лечения. В перечень регулярно назначаемых при этом лекарств вошли препараты для контроля судорог (карбамазепин, клоназепам, ламотриджин, топирамат и др.), пищевые добавки (фолат, креатинин, карнитин, орнитин и др.). Некоторым пациентам было назначено профилактическое лечение для снижения риска развития осложнений, специфичных для их заболеваний.
Почти четверть пациентов (22 %) после постановки диагноза направили на дополнительные обследования к врачам негенетических специальностей, чаще всего к кардиологам (28 %), а также к специалистам по нефрологии (13 %), офтальмологии (11 %), радиологии (10 %), неврологии (7 %), эндокринологии (7 %), аудиологии (3 %), стоматологии (2 %), дерматологии (2 %), ортопедии (1 %). Треть пациентов получили более одного направления к разным специалистам. У многих пациентов своевременное лечение дополнительных патологий позволило избежать развития тяжелых осложнений. Ученые также указывают, что 10 % пациентов могли избежать таких процедур, как МРТ и биопсия мышц, если бы диагноз был поставлен раньше.
Информационную поддержку по конкретному состоянию предоставили 3214 семьям (76%). В научных публикациях упоминаются 772 семьи (18 %). На момент сбора данных примерно с каждой третьей семьей обсудили возможности пренатальной диагностики или ЭКО с преимплантационным генетическим тестированием, но проводились эти процедуры только в 2 % случаев. Исследователи считают, что эти цифры были бы выше, если бы родители были моложе на момент постановки диагноза. Только 6 % родителей решились на повторную беременность после того, как узнали о генетическом заболевании своего ребенка. Исследователи также указывают, что 20,8 % семей, участвовавших в проекте, присоединились к группам поддержки пациентов, от небольших групп в соцсетях до крупных благотворительных организаций и фондов, и получили возможность общаться и обмениваться информацией с семьями, в которых есть аналогичные диагнозы.
«Примерно 1 из 17 человек страдает редким генетическим заболеванием, большинство из них — дети, и постановка правильного диагноза имеет решающее значение для получения наилучшего лечения и ухода, — говорит Кэролайн Райт, профессор геномной медицины в Университете Эксетера и корреспондирующий автор статьи. — Теперь у нас есть технология, позволяющая быстро поставить генетический диагноз примерно 50 % людей, страдающих редкими заболеваниями, но проблема может заключаться в том, достаточно ли данных для понимания конкретного заболевания, поскольку они так редки по отдельности».
Генетический анализ улучшил диагностику редких заболеваний на Среднем Востоке
Источники
Copeland H. et al. Large-scale evaluation of outcomes after a genetic diagnosis in children with severe developmental disorders // Genetics in Medicine Open. Published online October 14, 2024. DOI: 10.1016/j.gimo.2024.101864
Цитата по пресс-релизу

Вам будет интересно
 263
263
 0
0


 546
0
546
0