Меню
Меню





 Все темы
Все темы
Фаг Mushuvirus mushu практически не изменился за 1300 лет
Ученые из Польши и Германии использовали новейшие биоинформатические инструменты, чтобы повторно проанализировать данные, полученные при секвенировании ДНК из 150–5300-летних копролитов Европы и Северной Америки. Они выявили 298 предполагаемых древних вирусов кишечника человека. Один древний фаг из 1300-летнего образца был схож с современным фагом Mushuvirus mushu на 97,7%, что не соотносится с обычно довольно высокой скоростью мутации бактериофагов. Авторы выдвинули три гипотезы, которые могли бы объяснить этот феномен.

Современные методики позволяют изучать древние вирусы. Однако усилия исследователей в основном сосредоточены на патогенах человека, ретровирусных элементах и покоящихся вирусах эукариот. Очень мало известно о древних бактериофагах. Есть несколько посвященных им исследований, но в них нет данных о паттернах повреждений ДНК, отличающих древние нуклеиновые кислоты от современных. К тому же геномы древних бактериофагов были секвенированы только частично. В новом исследовании ученые из Польши и Германии проанализировали черновые и полные геномы из 150–5300-летних копролитов и идентифицировали древний бактериофаг, почти полностью идентичный современному виду Mushuvirus mushu, заражающему комменсальные бактерии.
Авторы выбрали ранее опубликованные наборы данных, полученные в результате секвенирования древней ДНК из 30 образцов кишечного микробиома человека. Образцы были отобраны в Европе и Северной Америке, их возраст оценивался в 150–5300 лет. В результате сборки de novo получили 72 693 контига высокого качества (средняя длина 12 592 нуклеотида). Однако, судя по паттерну дезаминирования 5’-конца, меньше половины из них принадлежали древней ДНК. Всего 2375 сиквенсов были признаны вирусными по крайней мере двумя методами. Из них 383 контига имели длину не менее 20 т.п.о. и качество выше среднего. В итоге было получено 298 вирусных оперативных таксономических единиц на уровне вида — это предполагаемые древние вирусы кишечника человека.
Чтобы исключить загрязнения ДНК из окружающей среды (например, из пещерных отложений), которая тоже могла подвергаться деградации, авторы предприняли дополнительные шаги. Они сравнили полученные данные с референсными вирусами из различных сред. Только 5% геномов предполагаемых древних вирусов кластеризовались с вирусами из окружающей среды. Один древний вирусный геном имел более чем 95%-е сходство с геномом современного фага M. mushu. Предполагается, что более половины древних вирусов заражало бактерии родов Clostridia и Bacteroidia, которые доминируют в кишечнике.
Из 298 древних вирусных последовательностей 293 отнесли к классу Caudoviricetes, три — к Megaviricetes, а две — к типам Cressdnaviricota и Phixviricota. Авторы выбрали 49 древних геномов высокого качества или полных древних геномов и проанализировали их отдельно. Их отнесли к 39 предполагаемым семействам и 46 предполагаемым родам.
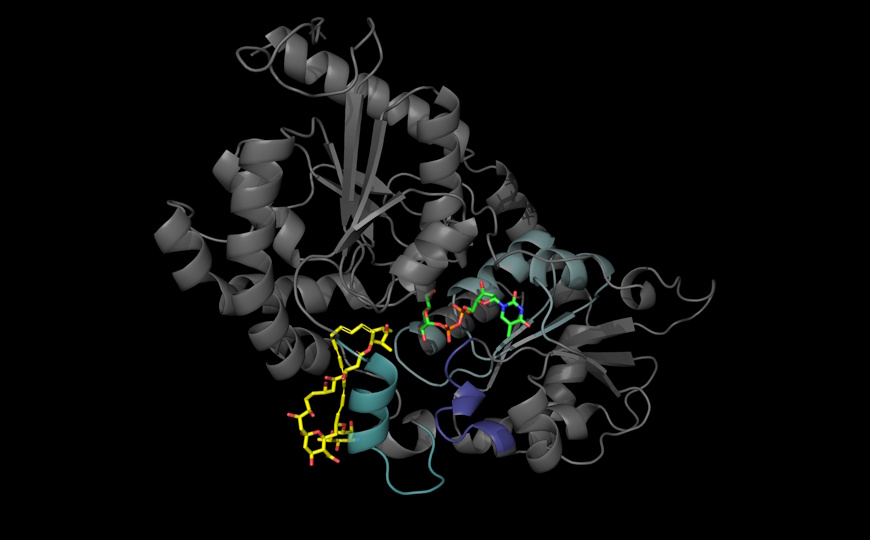
Хозяином M. mushu является Faecalibacterium prausnitzii, анаэробная бактерия, которая обычно обитает на слизистой оболочке желудочно-кишечного тракта. Древний геном был получен из мексиканского образца возрастом 1300 лет. Он на 97,7% был схож с референсным геномом FP_Mushu, который был извлечен из образца сточных вод во Франции. Древний и современный геномы имели приблизительно одинаковую длину (36 623 и 36 636 п.о.) и идеальную коллинеарность между 52 белок-кодирующими генами.
Таким образом, авторы использовали новейшие биоинформатические методы, чтобы воссоздать геномы древних фагов. Для валидации находок они проанализировали паттерны повреждения ДНК, связь с современными вирусами и с организмами-хозяевами. Обычно геномы бактериофагов быстро меняются под влиянием мутаций, рекомбинаций и горизонтального переноса генов. Тем удивительнее было видеть высокую консервативность древнего и современного геномов M. mushu. Авторы предложили три гипотезы, объясняющие, как это стало возможным.
Так, это может быть последствием стратегии репликации фага. При репликации M. mushu полагается на полимеразу III организма-хозяина, которая реже совершает ошибки, чем типичная фаговая полимераза. Вторая гипотеза связана с широким кругом хозяев M. mushu, куда входят многие представители Oscillospiraceae. Консервативность последовательности фага может быть связана с очищающим отбором. Третья гипотеза состоит в том, что фаг обычно существует в виде профага, интегрированного в геном бактерии, и изменяется одновременно с ним.
Источник:
Piotr Rozwalak, et al. Ultraconserved bacteriophage genome sequence identified in 1300-year-old human palaeofaeces // Nature Communications (2024), published 23 January 2024, DOI: 10.1038/s41467-023-44370-0




 0
0