Меню
Меню





 Все темы
Все темы
FDA модернизирует процедуру регистрации медицинских изделий
FDA планирует изменить процедуру для выдачи разрешений на продажу медицинских изделий, аналоги которых уже существуют на рынке.
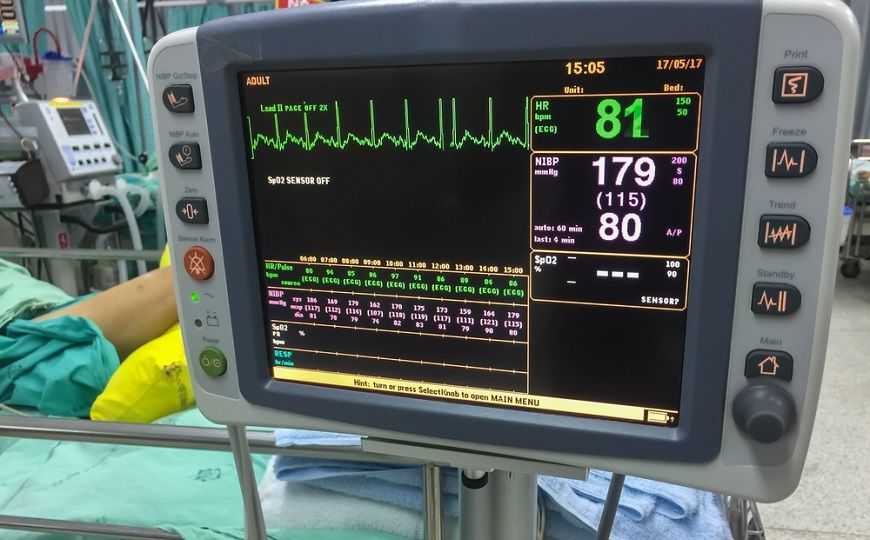
Разрешение на применение медицинских устройств на территории США выдает управление по контролю качества пищевых продуктов и лекарств (Food and Drug Administration, FDA) На сегодня FDA регулирует обращение более 190 000 медицинских изделий (далее медизделий). При регистрации новых изделий, кардинально отличающихся от существующих на рынке, проводятся серьезные испытания по определению эффективности и безопасности, по результатам которых FDA выносит решение. Процедура 510(k) — это упрощенный путь регистрации медизделий, аналоги которых уже существуют на рынке. FDA дает разрешение на продажу медизделия на основе сравнения с уже продающимися устройствами-аналогами; фактически производителю нужно только подтвердить, что новое устройство эквивалентно такому изделию.
Однако старые технологии могут не соответствовать современному пониманию рисков. Например, прикроватные системы мониторинга больных, информация с которых поступает на пост медицинской сестры, не защищены от кибератак. Старые устройства, использующие беспроводную связь, могут не иметь защиты от помех, связанных с появлением новых высокотехнологичных устройств. При этом производители новых медизделий могут использовать подобные устройства в качестве эталона для получения разрешения FDA, что, конечно, недопустимо.
26 ноября на сайте FDA появилась информация о том, что процедура 510(k) будет изменена и вновь выдаваемые разрешения уже не будут основываться только на сравнении с медизделиями, присутствующими на рынке. Закон, устанавливающий порядок оценки безопасности и эффективности медицинских устройств, был принят американским конгрессом 42 года назад, и чтобы идти в ногу с быстро развивающимися новыми технологиями, необходимы изменения. Анализ показал, что 20% регистрируемых сейчас медизделий сравниваются с аналогами, которым больше 10 лет. Модернизация 510(k) требует сравнения с современными аналогами, что будет способствовать развитию технологий здравоохранения, повышению безопасности регистрируемых медизделий. Устаревшие медизделия будут исключаться из числа действующих с появлением более безопасных или более эффективных технологий. Как заявляет FDA, новая процедура регистрации, сведет к минимуму риски при использовании медицинских устройств.
Источники

Вам будет интересно


 1020
1020
 0
0