Меню
Меню





 Все темы
Все темы
Интеграция данных пространственной транскриптомики и секвенирования РНК единичных клеток: цели и методы
Китайские исследователи сравнили 16 биоинформатических методов для интеграции данных пространственной транскриптомики (секвенирования РНК в точках на гистологическом срезе) и секвенирования РНК единичных клеток. Они выделили группы методов, лучше подходящие для прогнозирования пространственного распределения транскриптов, не выявленных напрямую, или для определения типов клеток по транскриптомным данным.
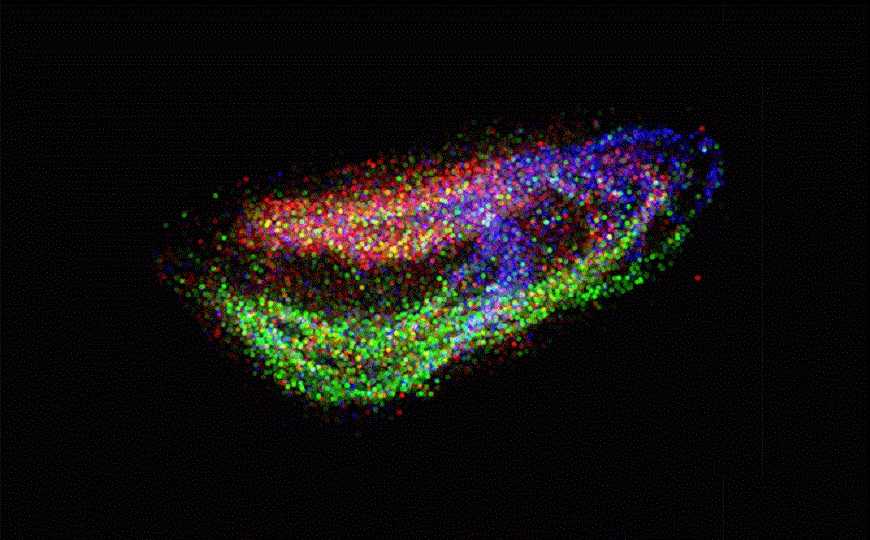
Трехмерная реконструкция девяти кубических миллиметров гиппокампа мыши, профилированная с помощью технологии пространственной транскриптомики Slide-seq
Credit:
Пространственные транскриптомные технологии обнаруживают транскрипты РНК в тканях. Методы, основанные на гибридизации in situ и флуоресцентной микроскопии (seqFISH, osmFISH, MERFISH), позволяют обнаруживать транскрипты с точностью до единичной клетки, но общее количество транскриптов, которое можно выявить, ограниченно. Подходы пространственной транскриптомики, основанные на секвенировании следующего поколения (NGS), такие как ST, 10X Visium, BGI Stereo-seq и Slide-seq, имеют противоположное ограничение — они исследуют весь транскриптом в той или иной точке, но каждая точка может содержать несколько клеток, то есть пространственное разрешение хуже, чем у визуальных методов.
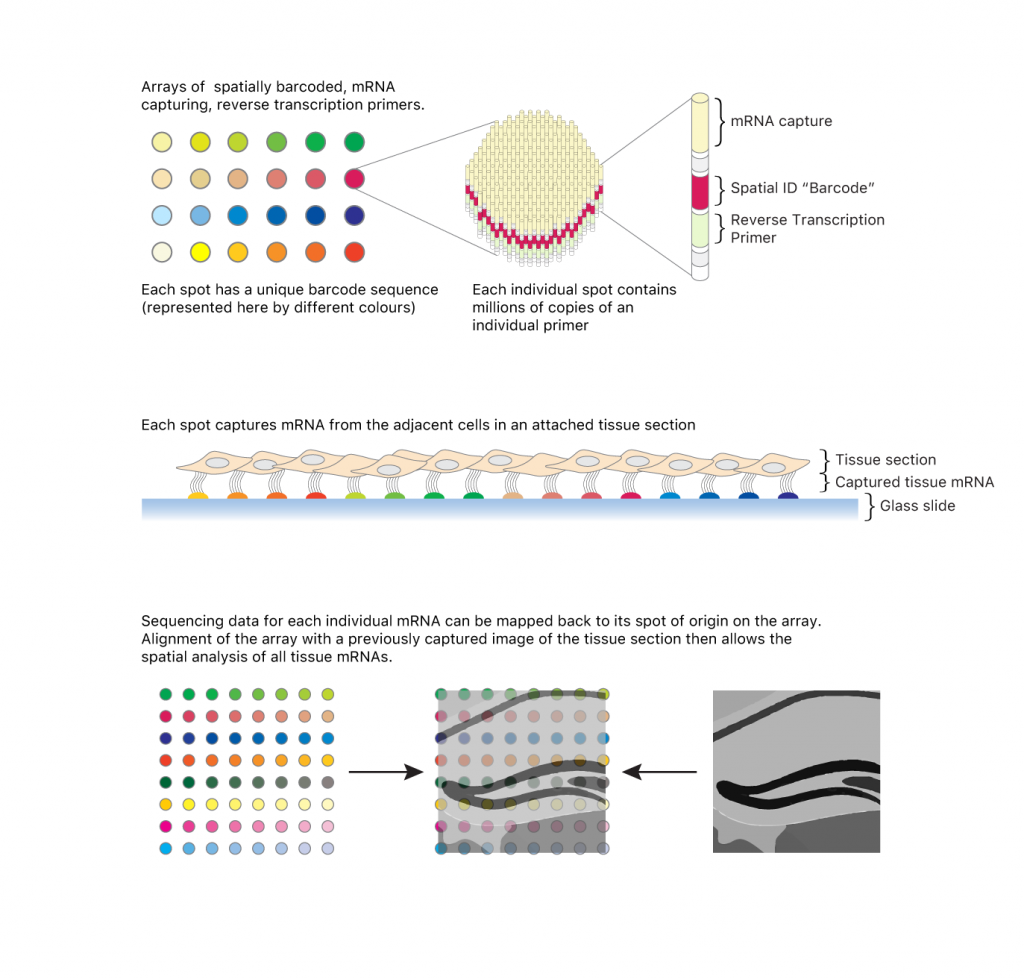 Принцип пространственной транскриптомики. James Chell | CC BY-SA 4.0
Принцип пространственной транскриптомики. James Chell | CC BY-SA 4.0
Чтобы преодолеть эти проблемы, создаются биоинформатические методы, объединяющие два типа данных — данные пространственной транскриптомики и секвенирования РНК единичных клеток (scRNA-seq; к ним относятся, например, Drop-seq, Smart-seq и 10X Chromium). Такие интегративные методы позволяют прогнозировать пространственное распределение транскриптов, не обнаруженных методами пространственной транскриптомики, и (или) выполнять деконволюцию типов клеток в точках на гистологических срезах. Однако до сих пор не было независимых попыток сравнить производительность различных методов интеграции.
Исследователи из Университета науки и технологии Китая выполнили сравнение 16 методов интеграции с использованием 45 парных наборов данных (данные пространственной транскриптомики и scRNA-seq) из опубликованных исследований и 32 смоделированных наборов данных. Они также оценили потребляемые вычислительные ресурсы.
По оценкам авторов, некоторые из методов (Tangram, gimVI, SpaGE)лучше подходят для предсказания пространственного распределения транскриптов РНК, в то время как другие (Cell2location, SpatialDWLS, RCTD) — для деконволюции клеточных типов в точках. Однако производительность метода в любом случае зависит от плотности матрицы экспрессии в методе пространственной транскриптомики.
«Наше исследование помогает ученым выбирать подходящие инструменты и оптимизировать рабочие процессы анализа данных для точной и эффективной интеграции данных пространственной транскриптомики с данными scRNA-seq», — пишут авторы.
Источник
Li, B., et al. Benchmarking spatial and single-cell transcriptomics integration methods for transcript distribution prediction and cell type deconvolution. Nature Methods (2022). DOI: 10.1038/s41592-022-01480-9

Вам будет интересно
 2090
2090
 0
0