Меню
Меню





 Все темы
Все темы
Исследование экзомов 30 тысяч семей указало на важность повторного анализа мутаций в генах, связанных с нарушениями развития
Многие нарушения развития связаны с рецессивными мутациями в определенных генах, однако количественной оценке вклада таких мутаций в развитие заболеваний пока посвящено не так много масштабных работ. Коллектив из США провел анализ рецессивных мутаций на большой и генетически разнородной группе, включающей около 30 тысяч триад «мать-отец-ребенок», и пришел к выводу, что повторный анализ уже известных генов, связанных с нарушениями развития, необходим для улучшения диагностики и интерпретации вариантов в этих генах.

Нарушения развития часто имеют генетическую природу — их могут вызывать аутосомно-рецессивные мутации в белок-кодирующих генах. Гены, мутации в которых ассоциированы с патологиями, называют генами ARDD (autosomal recessive developmental disorder-associated genes), и работы по их выявлению активно продолжаются. Команда исследователей из Института Сэнгера (Wellcome Sanger) в Великобритании в коллаборации с сотрудниками компании GeneDx из США, занимающейся коммерческими генетическими исследованиями, считает, что повторный анализ уже идентифицированных мутаций в известных генах ARDD улучшит диагностику нарушений развития во множестве семей по всему миру. Для проверки этой гипотезы они проанализировали полные экзомы 30 тысяч триад (в них входили мать, отец и ребенок с нарушением развития). Это в целых шесть раз больше, чем было в предыдущем исследовании авторов. В анализ включили экзомы людей африканского, американского, европейского, азиатского и ближневосточного происхождения.
Анализ рецессивных генетических вариантов в группах разного происхождения выявил, что от 2 до 19% случаев нарушения развития сопряжены с аутосомно-рецессивными мутациями. Причем такая вариабельность может быть связана с числом близкородственных связей в популяции — на это указывает корреляция между данными факторами.
Используя базы данных известных генов ARDD ( DDG2P и собственный список компании GeneDx), авторы показали, что мутации в таких генах объясняют около 80% случаев, описанных в исследуемых данных. Причем для европейской популяции эта доля оценивается в 87%, а для всех остальных — в 80%.
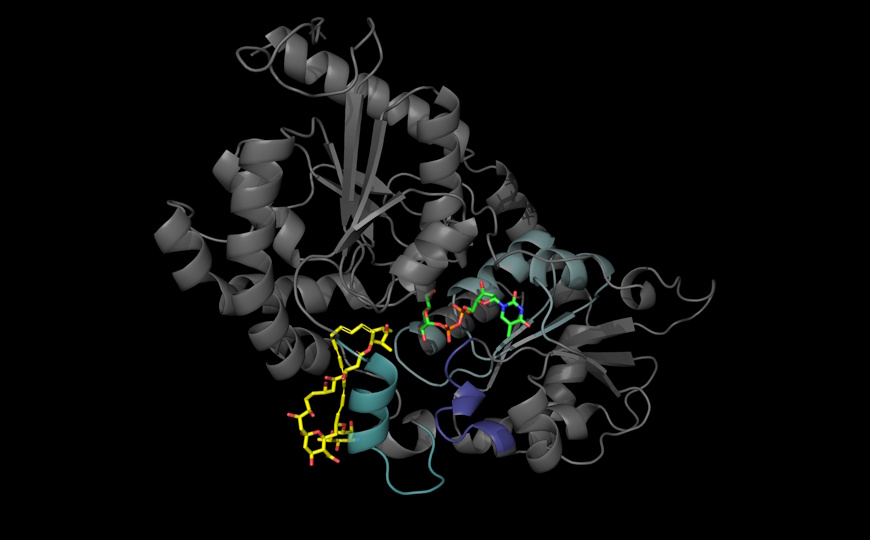
Несмотря на это, исследователи обнаружили пять новых потенциальных генов ARDD: CRELD1, KBTBD2, ZDHHC16, HECTD4 и ATAD2B. Вклад двух из них — KBTBD2 и CRELD1 — сохранял статистическую значимость после дополнительных поправок на множественное сравнение. KBTBD2 кодирует адаптер убиквитинлигазы и регулирует уровень p85α —субъединицы фосфоинозитол-3-киназы, ответственной за сигналинг инсулина. Его роль в фенотипические изменения была показана на мышах: его нокаут приводил к задержке роста, стеатозу печени и диабету. У обоих пациентов с мутациями в этом гене также наблюдались задержки роста, а у старшего еще и диабет.
Увеличение доли случаев нарушений развития, связанных с аутосомно-рецессивными мутациями, по сравнению с предыдущими исследованиями авторов отражает выявление новых генов ARDD за последние шесть лет. Однако высокий процент мутаций в уже известных генах ARDD в исследуемом датасете подчеркивает важность повторного анализа и интерпретации уже известных мутаций. Доктор Винсент Устач, старший автор исследования и сотрудник компании GeneDx, комментирует: «Это самая разнообразная группа участников, когда-либо изучавшаяся на предмет вклада рецессивных мутаций в нарушения развития, и она демонстрирует ключевое влияние разнообразия данных на достижение более полного понимания нарушений развития при различном происхождении». Авторы рассчитывают, что их выводы о необходимости повторного анализа мутаций поспособствуют получению более подробной информации и улучшат диагностику нарушений развития, в частности, в недопредставленных группах населения.
Ген ATG4D, возможно, связан с нарушениями развития нервной системы
Источник
Цитата по пресс-релизу

Вам будет интересно

 93
93
 0
0


 92
0
92
0
 112
0
112
0
 132
0
132
0