Меню
Меню





 Все темы
Все темы
Мутации в гене SPTBN1 вызывают неврологические нарушения
Исследовали из клиники Майо (США) и коллабораторы проанализировали сиквенсы 29 людей с задержкой в развитии речи и моторики, умственной отсталостью, судорогами и другими неврологическими нарушениями. Выявили 28 вариантов в гене SPTBN1, который кодирует белок βII-спектрин. Мутации в этом гене влияют на организацию цитоскелета, морфологию и взаимодействие нейронов.
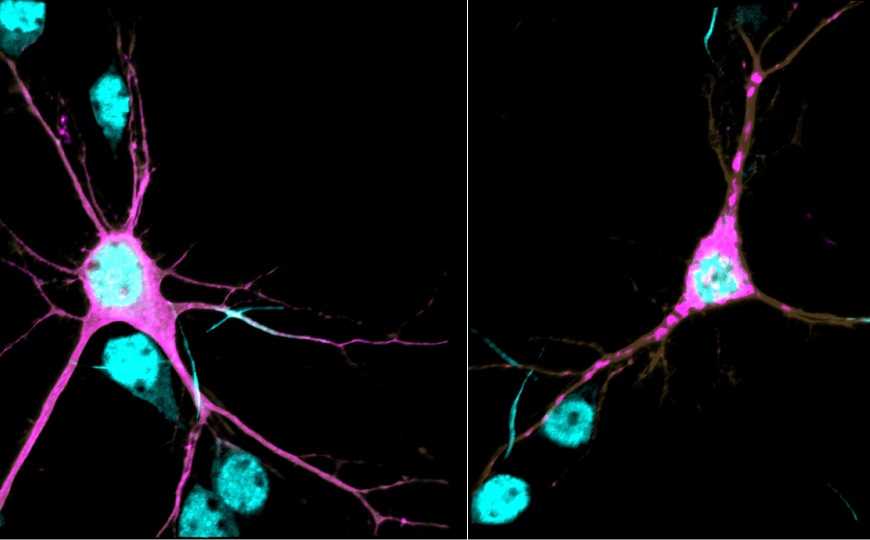
Lorenzo Lab, UNC School of Medicine | Пресс-релиз
Белки цитоскелета спектрины выстилают клеточную мембрану и обеспечивают ее стабилизацию и целостность. Дефицит спектринов приводит к различным нарушениям нервной системы: атаксиям, эпилептическим синдромам, невропатиям и множеству других расстройств. Ген SPTBN1 кодирует βII-спектрин, белок, который в большом количестве экспрессируется в головном мозге.
Исследовали из клиники Майо (США) и коллабораторы с помощью полногеномного или полноэкзомного секвенирования выявили 28 вариантов в гене SPTBN1 у 29 человек. В гетерозиготе эти варианты вызывали различные неврологические нарушения, включая задержку в развитии речи и моторики, умственную отсталость, расстройства аутистического спектра, судороги, двигательные аномалии, синдром дефицита внимания и гиперактивности. Примечательно, что у людей с одинаковыми или похожими генетическими вариациями наблюдались разные клинические проявления. Ученые полагают, что патогенные варианты гена SPTBN1 вызывают неврологический синдром с широким спектром проявлений.
Авторам исследования удалось выяснить молекулярный механизм патологического действия выявленных вариантов гена. Патогенные варианты дестабилизируют βII-спектрин, нарушают связывание белка с ключевыми молекулярными партнерами, а также влияют на организацию и динамику цитоскелета. Для изучения влияния патологических вариантов гена SPTBN1 на функцию белка ученые использовали молекулярные, клеточные и компьютерные модели, клетки человека и мыши, а также живых мышей.
Кроме того, эти варианты изменяют морфологию нейронов и их взаимодействие. Снижение уровня βII-спектрина в нейронах мутантных мышей нарушало структурную связь между кортикальными областями.
«Интересно, что разные варианты ведут себя совершенно по-разному, причем некоторые из них делают белок бета-два спектрин нестабильным, а некоторые нарушают его способность вступать в важные взаимодействия с другими белками», — говорит ведущий автор исследования Марго Кузен, исследователь трансляционной геномики в клинике Мэйо. — Но эти различия в эффектах помогли объяснить различия в клинических проявлениях, которые мы наблюдали у пациентов».
По мнению ученых, определения вариантов SPTBN1, лежащих в основе множества неврологических расстройств, является первым и очень важным шагом на пути к разработке терапии.
Источники
Cousin M.A., et al. // Pathogenic SPTBN1 variants cause an autosomal dominant neurodevelopmental syndrome // Nature Genetics, published July 1, 2021, DOI: 10.1038/s41588-021-00886-z
Цитата по пресс-релизу
Вам будет интересно


 215
215
 0
0