Меню
Меню





 Все темы
Все темы
Предложены эталоны для более точного определения медицински значимых вариантов генов
Новые эталонные последовательности, разработанные The Genome in a Bottle Consortium Национального института стандартов и технологий США, помогут идентифицировать многие сложные для выявления генные варианты, ассоциированные с болезнями.
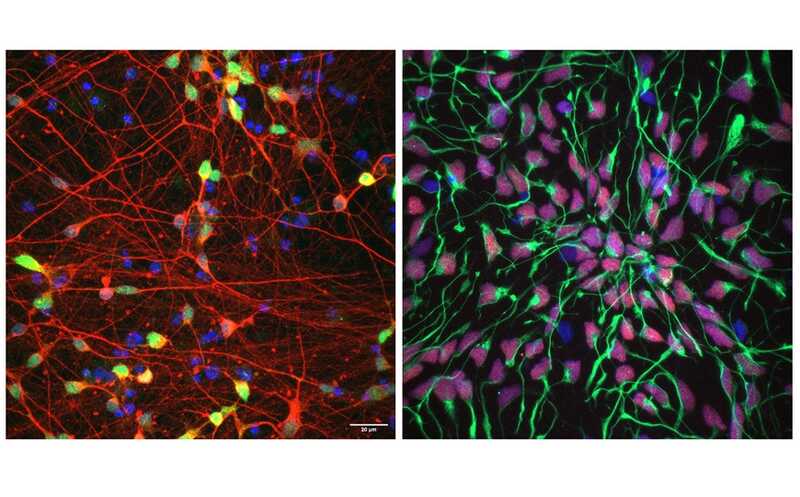
B. Hayes/NIST
Группа под руководством исследователей из Национального института стандартов и технологий (NIST) США, Медицинского колледжа Бэйлора и компании DNAnexus предоставляющей облачную платформу для анализа и управления данными о секвенировании ДНК, описали более 20 000 медицински значимых вариантов 273 генов.
The Genome in a Bottle Consortium (GIAB) — это государственно-частно-академический консорциум, организованный NIST для разработки технической инфраструктуры (справочные стандарты, эталонные методы, эталонные данные), которая поможет внедрить полногеномное секвенирование в клиническую практику. GIAB уже представил наборы эталонов для выявления однонуклеотидных вариаций (SNV), небольших инсерций и делеций (INDEL) и структурных вариантов (SV). (В данной работе INDEL — это вставки и делеции размером менее 50 п.н., а SV — вставки и делеции более 50 п.н.).
Эталоны — это последовательности ДНК, которые клиники и исследовательские лаборатории, тестирующие собственные методы секвенирования, могут использовать для самопроверки. Секвенировав тот же индивидуальный геном человека, который использовался для разработки эталона, а затем сравнив результат с самим эталоном, они поймут, насколько хорошо могут обнаруживать определенные варианты. В данной работе использовался геном конкретного индивида HG002; человек дал согласие на обнародование своей генетической информации в рамках Personal Genome Project.
В новой публикации представлен эталон для сложных и значимых с медицинской точки зрения генов (challenging, medically relevant genes CMRG). Это поможет лабораториям и клиникам более точно секвенировать эти гены, что имеет решающее значение как для диагностики, так и для исследований механизмов заболеваний и разработки терапии.
Недавнее исследование показало, что 13,8% (17 561) известных патогенных вариантов трудно обнаружить с помощью секвенирования коротких прочтений. Сложны для детекции, в частности, варианты размером 15–49 п.н., низкокопийные вариации числа копий (CNV), варианты в сегментарно дуплицированных областях. Тем не менее многие из этих вариантов возможно картировать с помощью высокоточных длинных прочтений.
Ключевую роль в новой работе сыграло секвенирование HiFi, которое позволяет получать риды длиной в десятки тысяч нуклеотидов. Также команда применяла hifiasm, программный инструмент, позволяющий исследовать отдельно «материнскую» и «отцовскую» копии генома человека, что исключает ошибки, вызванные гетерозиготности.
Чтобы определить приоритеты, авторы использовали несколько списков генов, потенциально значимых с медицинской точки зрения: список из 4773 генов, составленный в 2012 году на основе баз данных OMIM, HGMD и ClinVar, список COSMIC, содержащий 723 гена, ассоциированных с онкозаболеваниями, а также собственный список высокоприоритетных генов наследственных заболеваний, которые обычно тестируются в клинике. Итоговой список включал 5175 генов, 4697 из которых были аутосомными. Авторы сфокусировались на 395 «трудных» генах, которые были расшифрованы меньше чем на 90%. В итоге работа охватила более 20 000 вариантов в 273 генах, включая десятки трудно поддающихся оценке структурных вариантов.
«Некоторые из этих генов, к которым раньше было очень трудно получить доступ, подозреваются в какой-то ассоциации с болезнью. Другие имеют явную клиническую значимость, — сказал биомедицинский инженер NIST Джастин Зук, соавтор исследования. — Например, SMN1 — это охарактеризованный нами ген, который напрямую связан со спинальной мышечной атрофией — редким, но тяжелым заболеванием». Менее надежная сборка генов SMN1 и SMN2 может быть вызвана как раз проблемами с материнским и отцовским гаплотипами.
Помимо спинальной мышечной атрофии, исследователи охарактеризовали варианты в генах, связанных с сердечными заболеваниями, диабетом, чувствительностью к глютену и многими другими состояниями.
Команда также нашла ошибки в двух референсных геномах, которые они использовали ранее. По словам Зука, некоторые из ошибок могут помешать выявлению вариантов, связанных с серьезными заболеваниями, включая гомоцистинурию, которая связана с нарушениями скелета, сердечно-сосудистой и нервной систем и обычно выявляется при скрининге новорожденных.
Источник
Justin Wagner, Jet al. Curated variation benchmarks for challenging medically relevant autosomal genes // Nature biotechnology, Published: 07 February 2022; DOI: https://doi.org/10.1038/s41587-021-01158-1

Вам будет интересно
 263
263
 0
0


 546
0
546
0