Меню
Меню





 Все темы
Все темы
Синтез бета-амилоида в печени вызывает признаки болезни Альцгеймера у мышей
Австралийские ученые создали трансгенных мышей, синтезирующих бета-амилоид только в печени. На этой модели они показали, что бета-амилоид, который производится периферическими органами, вызывает патологические изменения в мозге, сходные с признаками болезни Альцгеймера.
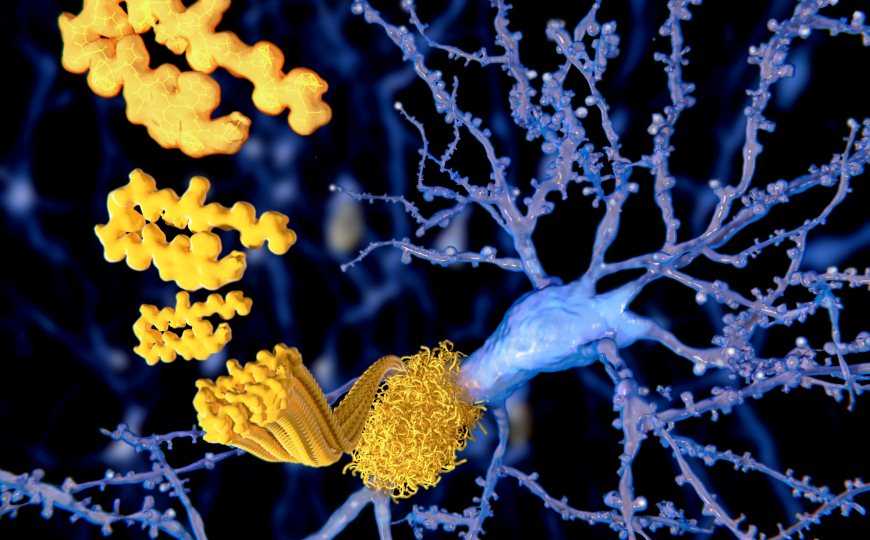
Накопление бляшек бета-амилоида (Aβ) в головном мозге — один из ключевых признаков болезни Альцгеймера (БА), приводящий к нейродегенерации как у людей, так и у модельных животных. Но бета-амилоиды также образуются в периферических органах. Уровни Aβ в крови коррелируют с церебральной амилоидной нагрузкой и снижением когнитивной функции, а значит, периферические бета-амилоиды могут способствовать заболеванию. В новой работе ученые показали на мышах, что синтез амилоидов в печени приводит к нарушениям, характерным для БА.
Более 90% Aβ в крови образуют комплекс с липопротеинами плазмы, которые состоят из белков (аполипопротеинов) и триглицеридов (TRL). Липопротеины низкой плотности (ЛПНП) переносят жиры из печени к периферическим органам, а липопротеины высокой плотности (ЛПВП) — наоборот. Высокий уровень ЛПНП связан с риском атеросклероза и других сосудистых патологий, ЛПВП же снижают этот риск. Бета-амилоиды используют липопротеины, чтобы перемещаться по организму. Причем чаще бета-амилоиды образуют комплексы с ЛПНП. Проникновение таких комплексов (в новой работе они обозначены как TRL-Aβ) в головной мозг может стать причиной церебральной амилоидной ангиопатии — повреждения сосудов мозга, которое способствует прогрессии БА.
Чтобы выяснить, как синтезированные вне мозга бета-амилоиды и метаболизм комплексов TRL-Aβ влияют на риск развития БА, авторы новой работы создали трансгенных мышей, экспрессирующих человеческий бета-амилоид только в гепатоцитах. Мыши получили название HSHA (hepatocyte-specific human amyloid).
На первом этапе ученые подтвердили, что значимо повышенная экспрессия человеческого амилоида наблюдается только в печени мышей HSHA. Однако, по данным ПЭТ, в мозге этих мышей происходит накопление амилоидов, причем сигнал становится сильнее с возрастом.
Кроме того, экспрессия печеночного APP (ген предшественника бета-амилоида) у мышей HSHA вызывала ускоренное накопление липидов в головном мозге, подобное таковому при БА. Наряду с этим у мышей HSHA уровень нейродегенерации (определялся с помощью окрашивания разрушенных нейронов в препаратах мозга) был примерно в два раза выше по сравнению с контрольными мышами в возрасте 6, 12 и 18 месяцев. Нейродегенерация была связана с повышенной проницаемостью капилляров в коре и гиппокампе. В норме капилляры становятся более проницаемыми с возрастом, но у трансгенных мышей это произошло несвоевременно. В мозге мышей HSHA детектировались признаки, характерные для БА, например, уменьшение объема гиппокампа и увеличение желудочков. Кроме того, трансгенные мыши плохо справлялись с тестом пассивного избегания, результаты которого зависят от работы гиппокампа.
Еще Алоис Альцгеймер описывал значительную инфильтрацию нейтральных липидов в образцах головного мозга пациентов с БА, однако это явление редко рассматривают в контексте этиологии заболевания. Авторы работы предполагают, что причиной нейродегенеративного фенотипа может быть метаболизм TRL-Aβ.
Исследование демонстрирует, что хроническое чрезмерное воздействие TRL-Aβ на сосуды ускоряет разрушение церебральных капилляров. Сосудистая патология в раннем возрасте повышает риски развития болезни Альцгеймера.
Ранее было показано, что у мышей дикого типа диета с высоким содержанием насыщенных жирных кислот приводит к усилению биосинтеза и секреции комплексов TRL-Aβ, их проникновению в мозг, повреждению капилляров и развитию нейроваскулярного воспаления. Диета, богатая ненасыщенными жирными кислотами, напротив, снижает количество TRL-Aβ.
Это означает, что диета или препараты, снижающие уровень TRL-Aβ в крови, могут уменьшать риск или замедлять прогрессирование БА.
Источник
Lam V., et al. Synthesis of human amyloid restricted to liver results in an Alzheimer disease–like neurodegenerative phenotype. // PLOS BIOLOGY, published September 14, 2021, DOI: 10.1371/journal.pbio.3001358



 0
0