Меню
Меню





 Все темы
Все темы
Векторная доставка гена микродистрофина для лечения миодистрофии Дюшенна
Ученые из США разработали метод генной терапии мышечной дистрофии Дюшенна (МДД) и проверяют его в клиническом исследовании, которое началось в ноябре 2017 года и продлится до марта 2021 года. Предварительные результаты опубликованы в журнале JAMA Neurology. У всех пациентов — участников исследования наблюдаются улучшения биологических и клинических показателей.
Миодистрофия Дюшенна (МДД) — это сцепленная с Х-хромосомой рецессивная болезнь, которую вызывают мутации в гене DMD, кодирующем дистрофин. У людей с МДД происходит дегенерация мышечных волокон, что приводит к раннeй инвалидизации и смерти.
Для некоторых болезней, вызванных мутациями, рассматриваются возможности генной терапии: функциональный ген доставляется в клетки пациента при помощи векторных конструкций на основе aденоассоциированных вирусов (AAV). Например, такую терапию успешно применили для лечения спинальной мышечной атрофии — заболевания, связанного с мутациями в генах SMN1 и SMN2.
Ген DMD — это самый большой ген в геноме человека, его длина составляет 2,4 Mb. Максимальная длина ДНК, которую можно доставлять с помощью AAV-векторов, составляет примерно 4,7 Kb, однако еще в 90-е годы 20 века было показано, что можно создать функциональный укороченный вариант дистрофина — микродистрофин.
Специалисты компании Sarepta Therapeutics разработали вектор для доставки гена микродистрофина в ядро человеческой клетки. Они использовали промотор MHCK7, который обеспечивает эффективную тканеспецифичную экспрессию этого гена в мышцах, а также капсид AAV, который отличается высоким тропизмом к клеткам мышц. Эта генноинженерная конструкция получила название rAAVrh74.MHCK7.micro-dystrophin (SRP-9001).
В клиническом исследовании SRP-9001 приняли участие четыре пациента — мальчики 4–7 лет. Препарат вводился пациентам однократно, внутривенно.
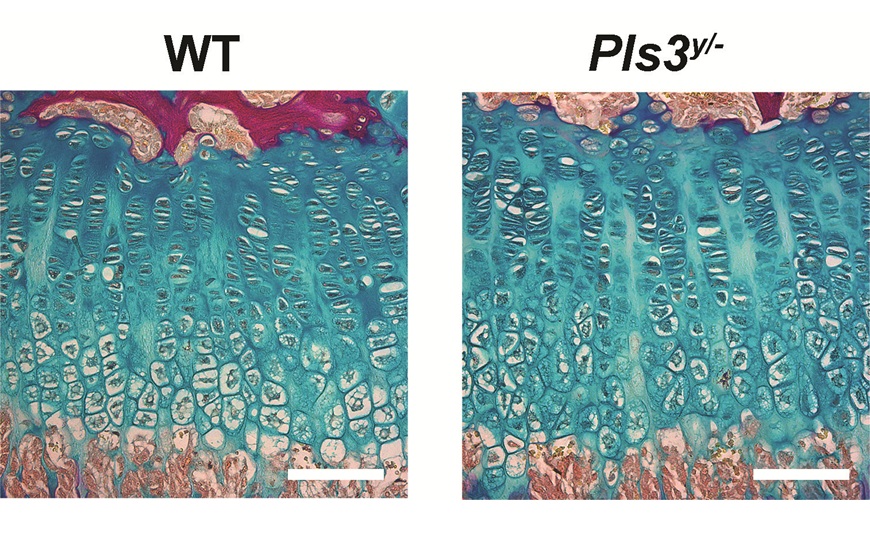
Биопсии мышечных тканей пациентов забирали до начала исследования и через 90 дней после введения препарата. Ученые использовали их для гистологического анализа, а также для проверки экспрессии микродистрофина методом иммуноблоттинга.
Серьезных побочных эффектов от введения препарата не наблюдалось. После инфузии SRP-9001 в мышцах детектировался микродистрофин. На основе анализа биопсий авторы делают вывод, что введение гена микродистрофина приводит к восстановлению функций белкового комплекса, ассоциированного с дистрофином.
Динамику мышечной функции пациентов оценивали по шкале NSAA. Через год после введения препарата у всех мальчиков наблюдались улучшения моторики. У пациентов также детектировалось значительное снижение уровня креатинкиназы в крови. Данная работа послужила основанием для дальнейших испытаний SRP-9001 на большей выборке больных. В настоящее время проходит рандомизированное плацебо-контролируемое исследование препарата с участием 41 пациента.
«На данный момент миодистрофия Дюшенна остается смертельной болезнью, и, как правило, пациенты не живут дольше 20–30 лет. Это заболевание сложно лечить, и генная терапия предоставляет нам долгожданные возможности постараться изменить течение этой болезни», — говорит доктор Джерри Мендел, первый автор статьи и главный исследователь Центра генной терапии при Общенациональной детской больнице.Источник
Mendell J. R., et al. // Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in Children With Duchenne Muscular Dystrophy A Nonrandomized Controlled Trial. // JAMA Neurology, June 15, 2020; DOi: 10.1001/jamaneurol.2020.1484
Цитата по пресс-релизу


 0
0