Меню
Меню





 Все темы
Все темы
Беззащитные, или Что такое тяжелый комбинированный иммунодефицит
Существует группа наследственных заболеваний, связанных с мутациями в различных генах, которые приводят к одному результату — нарушению работы иммунной системы. Их объединяет общее название «тяжелый комбинированный иммунодефицит» (ТКИД). Разбираемся, что представляют собой эти заболевания, чем можно помочь больным сегодня и как продвигается разработка генной терапии.

Что такое ТКИД?
Система иммунитета защищает нас от всего чужеродного: вирусов, бактерий, паразитов, инородных тел. Без нее мы все равно что без радиационной защиты в центре атомного реактора, где вместо жесткого излучения — окружающие нас микроорганизмы. Конечно, поле деятельности системы иммунитета гораздо шире, чем защита от патогенов. Но сегодня мы сосредоточимся на защитной функции, так как именно ее недостаток выступает на первый план при тяжелом комбинированном иммунодефиците.
Так сложилось в медицине, что редкие заболевания с похожими патогенезом и проявлениями стараются объединить в группы. Это существенно облегчает подход к их диагностике и лечению, потому что позволяет переносить опыт и результаты исследований одного заболевания на другое. Врач, впервые столкнувшийся с новым заболеванием, но имевший опыт работы с похожими диагнозами, знает, какие шаги ему предпринять, чтобы правильно определить заболевание и помочь пациенту. Когда болезнь встречается всего у одного человека из миллиона, это очень важно.
К числу таких состояний относятся и тяжелые комбинированные иммунодефициты (ТКИД, англ. SCID — Severe Combined Immunodeficiency) — группа наследственных заболеваний, в основе которых лежит генетический дефект, нарушающий работу системы иммунитета. Все они характеризуются крайне тяжелым течением и постоянными инфекциями начиная с первого года жизни. В лучшем случае тяжелый комбинированный иммунодефицит приводит к постоянной изоляции ребенка и частым госпитализациям. В худшем — к летальному исходу.
Общая распространенность ТКИД составляет примерно один на 50000 новорожденных. Сегодня основной метод лечения этого заболевания — трансплантация костного мозга. В условиях дефицита и сложности подбора доноров большинство пациентов лишены возможности пройти эту процедуру. Давайте разбираться, что обуславливает трагичность тяжелых комбинированных иммунодефицитов, и как медицинское сообщество борется за жизнь таких больных.
Чем различаются разные формы ТКИД?
Долгий путь клеток иммунной системы начинается в красном костном мозге. Здесь стволовые клетки крови делятся на два ростка (линии). Из первого образуются эритроциты, тромбоциты, а также гранулоциты и моноциты, которые являются компонентами системы иммунитета. Из второго развиваются остальные представители иммунных клеток — Т- и B-лимфоциты, а также клетки с запоминающимся названием «натуральные киллеры» (NK-клетки).
Уже на этом этапе изучения системы иммунитета возникает вопрос: «Зачем же ей так много клеток?» Все дело в том, что они никогда не работают поодиночке. Одни клетки находят патоген, другие захватывают его, третьи уничтожают, четвертые запоминают, чтобы при повторной встрече уже знать, как с ним бороться. Сбой в любом из этих последовательных действий приводит к неспособности иммунной системы качественно отреагировать на внешнее воздействие. Именно в этих нарушениях кроются причины всех врожденных иммунодефицитов.
Сегодня мы сосредоточимся на тяжелых комбинированных иммунодефицитах. Для этих заболеваний характерны нарушения в работе Т-, B- и NK-клеток.
Т-хелперы (они же CD4+ Т-лимфоциты) помогают бороться с инфекцией Т-киллерам (CD8+ Т-лимфоцитам) — цитотоксическим Т-клеткам, которые убивают зараженные клетки, а также В-лимфоцитам, производящим антитела. NK-клетки аналогичны по функциям Т-киллерам, но относятся к врожденному иммунитету и распознают зараженные клетки быстро, на ее раннем этапе
Чтобы лучше разобраться в патогенезе тяжелого комбинированного иммунодефицита, рассмотрим наиболее частый путь развития иммунной реакции с участием лимфоцитов.
Начнем с проникновения чужеродного агента в организм. Пусть это будет бактерия. Ее поглощают специальные клетки, которые называются дендритными — «ветвящимися» — из-за большого количества отростков. (С отростками-дендритами нервных клеток это название никак не связано.) Поймав бактерию, дендритная клетка выставляет на своей поверхности ее антигены — чужеродные вещества — и привлекает Т-хелперы с помощью цитокинов (cyto — клетка, kinesis — движение). Это вещества белковой природы, которые взаимодействуя с рецепторами на поверхности клетки-мишени, передают ей информацию о том, что она должна делать: куда-то двигаться, делиться, синтезировать и выделять вещества или погибнуть.
У людей с тяжелым комбинированным иммунодефицитом могут быть мутации, которые приводят к дефициту различных цитокинов или отсутствию на поверхности Т-хелперов рецепторов, принимающих их сигналы.
Например, мутации в гене субъединицы рецепторов интерлейкинов IL2RG препятствуют появлению зрелых иммунных клеток. Интерлейкины (разновидность цитокинов) необходимы для их развития. Без рецепторов клетки не могут принимать интерлейкиновые сигналы, и у пациента нарушается формирование Т-клеток и NK-клеток, а без Т-хелперов и В-клетки не работают должным образом. Ген IL2RG находится в Х-хромосоме, и многочисленные мутации в нем вызывают Х-сцепленный ТКИД, который чаще встречается у мальчиков. Это наиболее распространенный тип ТКИД.
Но предположим, что на этом этапе все прошло хорошо и Т-хелперы добрались до дендритных клеток. Первым делом они должны распознать, какой антиген им показывают. Количество чужеродных веществ в окружающей нас среде превышает количество клеток системы иммунитета на несколько порядков. Чтобы распознать это громадное разнообразие антигенов, на поверхности Т- и B-лимфоцитов есть специальные рецепторы. От других белков нашего организма они отличаются тем, что в их генах при созревании клетки иммунной системы происходят многочисленные перестройки. В результате этих перестроек образуются новые аминокислотные последовательности в антигенсвязывающих областях рецепторов. Они настолько разнообразны и представлены в таком большом количестве, что позволяют распознать антигены почти всех патогенов, включая бактерии, вирусы, паразитов и червей, а также измененные собственные клетки.
При тяжелом комбинированном иммунодефиците процессы перестройки в генах рецепторов могут нарушаться, что приводит к неспособности лимфоцитов распознать чужеродные вещества.
Распознав антиген как чужеродный, Т-хелперы привлекают цитотоксические (убивающие зараженные клетки) Т-лимфоциты или B-лимфоциты, продуцирующие антитела, которые связываются с антигенами. Антитела на поверхности микроорганизма служат ориентиром для моноцитов, чтобы те могли поглотить и переработать потенциально опасных гостей. В любом случае история закончится уничтожением патогена. Но только при условии, что все разновидности клетки иммунной системы присутствуют в достаточном количестве.
Если у человека не формируются лимфоциты, ему просто нечем защититься от инфекции. Мутации в гене ADA, кодирующем аденозиндезаминазу (АДА), могут приводить к накоплению токсичных продуктов метаболизма в клетках-предшественницах лимфоцитов и их гибели. АДА-ассоциированный ТКИД — второй по распространенности после Х-сцепленного, вызванного мутациями в IL2RG.
Интересно, что нет ни одного тяжелого комбинированного иммунодефицита с аутосомно-доминантным типом наследования. Для всех заболеваний характерно аутосомно-рецессивное или X-сцепленное наследование (болезнь проявляется только в том случае, если дефектный ген унаследован от обоих родителей или находится в Х-хромосоме у мальчика).
Теперь мы знаем большинство механизмов развития тяжелого комбинированного иммунодефицита. Давайте посмотрим, как он проявляется.
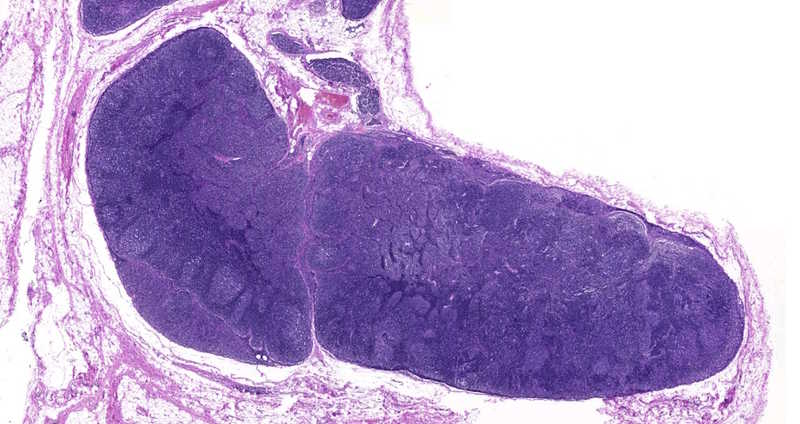
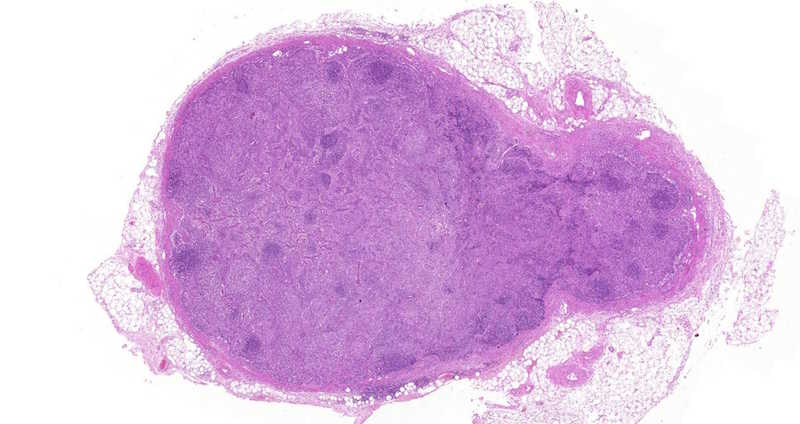 Микрофотографии лимфоузлов с нормальным гистологическим строением (вверху) и с морфологической картиной тяжелого иммунодефицита (внизу). Основные различия, которые можно отметить, — резкое уменьшение количества лимфоидных фолликулов. Изображения любезно предоставил Д.В. Барам
Микрофотографии лимфоузлов с нормальным гистологическим строением (вверху) и с морфологической картиной тяжелого иммунодефицита (внизу). Основные различия, которые можно отметить, — резкое уменьшение количества лимфоидных фолликулов. Изображения любезно предоставил Д.В. Барам
Как проявляется заболевание?
Считается, что в течение первых месяцев жизни основную роль в защите ребенка выполняют материнские антитела. Все заболевания из группы тяжелых комбинированных иммунодефицитов относятся к врожденным состояниям, но их проявления чаще всего начинаются с 2-6 месяцев, когда запас материнских антител истощается. Родители замечают, что их ребенок часто болеет. И не простудными заболеваниями, которые переносят все дети, а другими инфекциями, гораздо более тяжелыми. Это могут быть кишечные инфекции с упорной диареей, инфекции кожи и слизистых, вызванные грибками рода Candida. Наиболее опасны поражения респираторного тракта. К ним могут привести даже микроорганизмы, которые в норме не патогенны для человека, например, пневмоцисты.
Заподозрив тяжелый комбинированный иммунодефицит, врач первым делом должен проверить, соответствует ли клиническая и лабораторная картина критериям этого заболевания. В них оценивается наличие тяжелых инфекций, семейный анамнез, возраст развития проявлений, количество различных фенотипов Т-лимфоцитов в крови и маркеры их созревания. Также врачу необходимо исключить заражение ребенка вирусом иммунодефицита человека, который может передаваться от инфицированной матери.
Если ребенок соответствует диагностическим критериям, начинается поиск мутаций, которые стали причиной этого состояния. Проводится молекулярно-генетическое исследование, то есть анализ ДНК, выделенной из крови младенца. Для этого существуют различные методы. Генетические панели обеспечивают секвенирование («прочтение») только генов, связанных с развитием иммунодефицита. Полноэкзомное секвенирование — определение последовательность всех генов, которые кодируют белки; этот подход дороже, но он позволяет найти больше мутаций, которые могут служить причиной заболевания.
После истощения пула материнских антител дети с тяжелым комбинированным иммунодефицитом остаются беззащитными перед любой инфекцией. Поэтому первой мерой становится изоляция. Ребенка отлучают от материнской груди и помещают в специальный стерильный бокс, куда проникает только полностью очищенный от микроорганизмов воздух. Кормят таких детей стерилизованными смесями. Инъекции и другие манипуляции проводят с помощью специальных вмонтированных рукавов с перчатками.
Вылечив острое состояние, детей возвращают в мир. Но строгие правила остаются. Ребенку с установленным диагнозом тяжелого комбинированного иммунодефицита приходятся избегать контакта с другими людьми и животными. Гулять можно только летом, в детский садик и школу ходить нельзя. Мир ребенка ограничивается квартирой и родителями, которым, кстати, тоже приходится быть намного осторожнее, чтобы не принести домой инфекцию.
Кроме того, ребенку мешают наслаждаться детством частые госпитализации. Они могут проводиться как в плановом порядке, так и в экстренном, потому что какие бы меры ни предпринимали родители, ребенок рано или поздно заболеет. Такие дети с самого раннего возраста знают, что такое больницы, и понимают хрупкость своей жизни.
Широкий круг читателей узнал о тяжелых генетических заболеваниях, нарушающих работу иммунной системы, благодаря «мальчику в пузыре» Дэвиду Веттеру (1971–1984). У Дэвида был Х-сцепленный ТКИД — дефектный ген IL2RG в Х-хромосоме. Как мы помним, в этом случае у человека не созревают клетки иммунной системы. Свою короткую жизнь ребенок прожил в стерильном коконе, который покидал всего несколько раз в скафандре, подаренном NASA. Предполагалось, что он проведет в пузыре немного времени, а затем ему пересадят костный мозг, взятый от его сестры Кэтрин, но оказалось, что она не вполне подходящий донор. На поддержку его жизни было потрачено около 1,3 млн долларов. В октябре 1983 года Дэвиду все же трансплантировали костный мозг Кэтрин, так как полностью совместимого донора найти не смогли. Операция прошла благополучно, однако в феврале 1984 года ребенок умер от лимфомы Беркитта, вызванной вирусом Эпштейна — Барр, который не был замечен в клетках трансплантата. У большинства людей инфекция протекает бессимптомно, но не тогда, когда иммунная система бездействует.
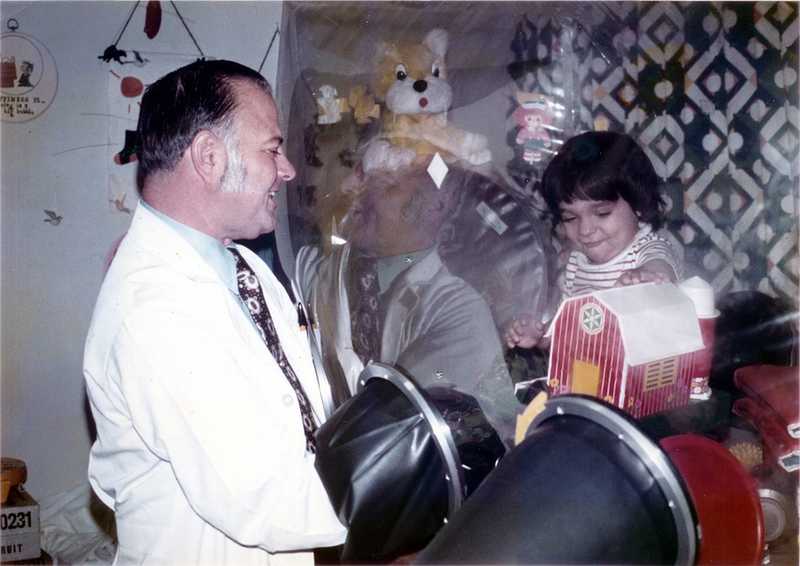 Дэвид Веттер и его врач Джон Монтгомери. Credit: Wikipedia
Дэвид Веттер и его врач Джон Монтгомери. Credit: Wikipedia
Существует ли терапия?
Как и для большинства редких заболеваний, используемых подходов терапии тяжелых комбинированных дефицитов не так много. И сегодня, помимо лечения инфекций с помощью антимикробных препаратов, каждый пациент рассматривается как потенциальный реципиент для пересадки костного мозга. Суть манипуляции заключается в уничтожении предшественников лимфоцитов с мутациями и заселение костного мозга новыми здоровыми клетками. Такой подход показал высокую эффективность (90%) для большинства заболеваний из этой группы, но ограничением остается дефицит подходящих доноров.
Для решения этой проблемы государства и благотворительные организации создают регистры доноров костного мозга. Каждый человек при отсутствии противопоказаний может пожертвовать свой костный мозг для помощи больным с такими тяжелыми заболеваниями, как ТКИД, лейкозы, апластическая анемия и др. Чем обширнее регистр, тем больше вероятность, что донор для конкретного пациента будет найден своевременно.
Разные заболевания, входящие в группу тяжелых комбинированных иммунодефицитов, являются результатом различных мутаций. Поэтому еще с конца ХХ века врачи-исследователи стараются подобрать терапию, которая исправит работу дефектного гена или возместит недостаток его продукта.
Препарат элапегадемаза (торговое название Revcovi, производитель — компания Chiesi Farmaceutici) применяется при ТКИД, вызванном мутациями в гене ADA. Это средство заместительной терапии содержат рекомбинантный белок, аналогичный человеческому ферменту аденозиндезаминазе, и вводится внутримышечно один раз в две недели. Дозировка подбирается по весу пациента. Согласно клиническим исследованиям, он восстанавливает количества Т и B лимфоцитов, а также NK-клеток у половины пациентов. К сожалению, он недоступен в РФ (только пациенты детского возраста имеют возможность получить его через фонд «Круг добра») и очень дорого стоит. Цена одной инъекции для ребенка весом 10 килограммов превышает 2 млн рублей, а инъекции нужны постоянно.
Альтернативой могут стать генотерапевтические препараты. Кстати, первым в мире человеком, получившим генную терапию, стала Ашанти Де Сильва с ADA-ассоциированным ТКИД. В 1990 году, когда Ашанти было четыре года, ее родители дали согласие на экспериментальную терапию — в Т-клетки девочки ввели «здоровый» ген с помощью ретровируса, и ее состояние улучшилось. (Правда, сообщается, что у второй пациентки, получившей то же лечение, измененные Т-клетки не смогли сформировать стабильную популяцию.) Тяжелых побочных эффектов у Ашанти не наблюдалось, она выросла и стала юристом по медицинским вопросам.
На первых этапах развития генной терапии многие попытки завершались неудачами или трагедиями. Только в 2016 году Европейское медицинское агентство одобрило первый генотерапевтический препарат Стримвелис для лечения АДА-дефицитов. Лечение было предназначено для пациентов, которым не удается подобрать донора костного мозга. Исследователи компании GlaxoSmithKline предложили брать собственные стволовые клетки крови пациентов, затем с помощью ретровирусного вектора вводить в них здоровый ген ADA и возвращать их обратно в организм.
Результаты клинических исследований подтвердили, что совершился прорыв: у всех пациентов после единственной процедуры полностью восстановились количества Т и B лимфоцитов, а также NK-клеток. Средняя частота тяжелых инфекций снизилась более чем в 6,5 раз.
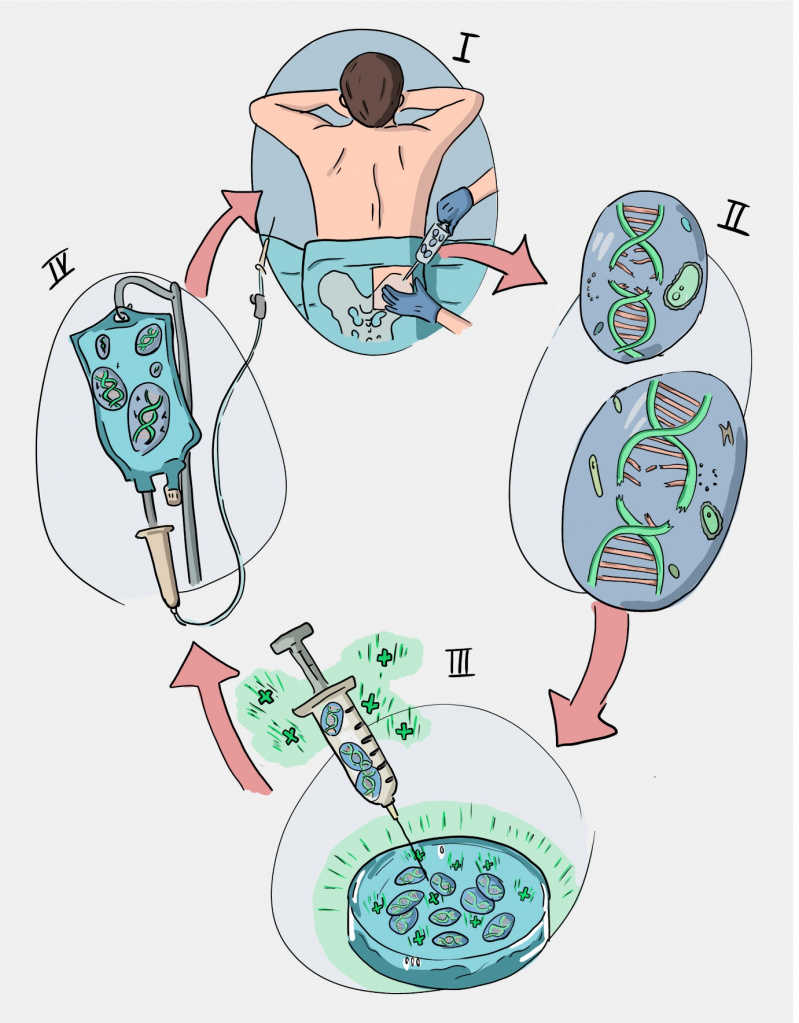 Механизм действия препарата Стримвелис. I — забор стволовых клеток крови из костного мозга пациента; II — схематичное изображение клеток с поврежденным геном ADA; III — культивирование стволовых клеток и трансдуцирование (заражение) их ретровирусным вектором, несущим здоровый ген ADA; IV — внутривенное вливание культивированных клеток. Художник: Владислав Ефремов
Механизм действия препарата Стримвелис. I — забор стволовых клеток крови из костного мозга пациента; II — схематичное изображение клеток с поврежденным геном ADA; III — культивирование стволовых клеток и трансдуцирование (заражение) их ретровирусным вектором, несущим здоровый ген ADA; IV — внутривенное вливание культивированных клеток. Художник: Владислав Ефремов
Но в 2019 году, через 4,7 года после процедуры, у одного из пациентов развился Т-клеточный лейкоз. В лейкемических клетках присутствовал здоровый ген ADA, поэтому врачи связали это нежелательное явление с терапией и инсерцией гамма-ретровирусного вектора в геном. Однако даже несмотря на это, ЕМА не отозвало разрешение на использование препарата. Ведь дети с тяжелым комбинированным иммунодефицитом часто умирают еще до двух лет, а участники испытания прожили больше пяти.
Исследовательские центры и биотехнологические компании продолжили исследования, чтобы сделать терапию модифицированными Т-клетками безопаснее. Ученые из США и Великобритании разработали подход с использованием самоинактивирующегося лентивирусного вектора со слабым промотором. При этом лентивирус встраивается в другие участки генома и за счет самоинактивации обладает меньшей транскрипционной активностью, что снижает его генотоксичность и онкогенный потенциал. В общей сложности 50 человек получили лечение этим препарата, который на этапе разработки назвали OTL-101. Он показал хороший профиль безопасности. За 36 месяцев наблюдения только 12 пациентов имели серьезные нежелательные явления, но все они были связаны с инфекциями или желудочно-кишечными заболеваниями. У всех пациентов была отмечена нормализация уровня ADA и достижение целевых возрастных уровней лимфоцитов.
После такого успеха были начаты исследования для других заболеваний из группы тяжелых комбинированных иммунодефицитов. Все они сосредоточены на доставке здоровых генов, которые у пациентов повреждены мутациями. Например, в конце 2023 года было закончено исследование трансдукции стволовых клеток с помощью ретровирусного вектора, несущего ген IL2RG. Окончательные результаты еще не опубликованы, но предварительный отчет показал полное восстановление Т лимфоцитов после лечения.
Несмотря на сложность и высокую стоимость разработки генной терапии, а также опасения, связанные с безопасностью, мы видим все больше примеров, показывающих эффективность такого подхода. Течение заболеваний, которые приводили к гибели или ранней инвалидизации пациентов, сейчас становится возможным контролировать, и терапия АДА-дефицита — один из ярких тому примеров.



 0
0