Меню
Меню





 Все темы
Все темы
Об успехах популяционной генетики и современной цитогенетике
Продолжаем рассказывать о докладах с пленарного заседания XI Съезда Российского общества медицинских генетиков (РОМГ). Как связана популяционная генетика с медицинской генетикой, почему у северных народов могут встречаться с 50%-ной частотой варианты генов, считающиеся патогенными, и в чем классическая цитогенетика превосходит секвенирование.

Вадим Степанов: «Не может в популяции быть 50% носителей патогенного варианта»
Академик РАН Вадим Анатольевич Степанов, директор Томского научно-исследовательского медицинского центра (НИМЦ), выступил с докладом о популяционной геномике человека. Этот раздел генетики занимается широкомасштабным изучением микроэволюции популяций человека. Секвенирование полных геномов в популяциях — технологическая основа данного направления — крайне важно для медицинской генетики и генетики болезней.
Полногеномная эра, в которую мы живем сейчас, характеризуется переходом к анализу индивидуальных геномов. Достижения этого направления хорошо известны — секвенирование генома человека и получение первого персонального генома в 2007 году, затем геномные проекты, такие как HapMap («карта гаплотипа», представляющая собой каталог частых SNP 11 популяций мира. —PCR.NEWS) и «1000 геномов». Все это сопровождалось активной трансляцией данных геномики в клиническую практику. Выявление вариантов, связанных с моногенными болезнями, полноэкзомное и полногеномное секвенирование входят в рутинную клиническую практику, в том числе в качестве тестов первой линии.
Однако зачем нужно секвенировать полные геномы? Это важно для нескольких направлений — популяционной геномики, медицинской генетики в узком смысле (в первую очередь генетики наследственных болезней и предиктивной медицины), фундаментальных эволюционных исследований. Более прикладные применения — создание баз данных по частотам патогенных вариантов и фармакогенетически значимых вариантов, криминалистика и судебная экспертиза, а также ДНК-идентификация и поиск родственников конкретного человека.
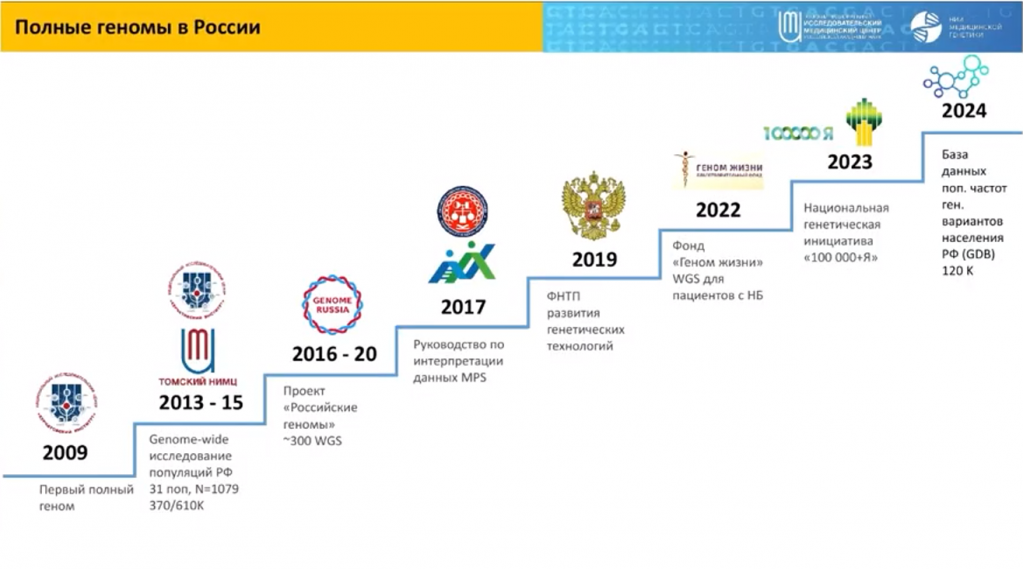
После проекта «1000 геномов» во многих странах, в том числе в России, сформировались национальные проекты, или геномные проекты второго поколения, задачей которых была каталогизация генетического разнообразия на уровне полных геномов в конкретной группе населения. Однако эти проекты были относительно небольшими, и они не были нацелены на решение задач медицины или здравоохранения. За ними стали появляться крупные проекты третьего поколения, некоторые из них реализуются и сейчас — Genomics England, GenomeAsia 100K и многие другие. Их принципиальное отличие от проектов предыдущего поколения заключается в масштабах — они на порядок или два крупнее, то есть золотым стандартом для такого типа проектов считается не менее ста тысяч геномов. Кроме того, все они обязательно имеют целью использование геномных данных для задач практической медицины.
История геномики в России тоже очень долгая и непростая, отметил докладчик — секвенирование первого полного генома в Курчатовском институте в 2009 году, затем несколько популяционных проектов, проект «Российские геномы», который, к сожалению, не очень удачно реализовался. Затем был сформирован фонд «Геном жизни», который дает возможность секвенировать геномы и назначать на основании этих данных препараты, необходимые пациентам с орфанными болезнями и зачастую дорогостоящие. Появление национальной генетической инициативы «100 000 + Я» также дает уникальный ресурс, который дополняет база данных вариантов, встречающихся у жителей РФ, — она получена на основании секвенирования 120 тысяч полных геномов.
Затем докладчик перешел к рассказу о собственных результатах, которые были получены в рамках двух довольно больших проектов. Первый — это научно-технический проект программы Союзного государства, реализованный с 2017 по 2021 годы консорциумом из нескольких российских учреждений, включая Томский МГНЦ, Институт общей генетики имени Н.И. Вавилова, и белорусских коллег. Проект был посвящен ДНК-идентификации, и в рамках этой программы было охарактеризовано более 100 популяций в России и Белоруссии — по более 20 тысяч образцов исследователи собрали несколько сотен полных геномов.
Второй крупный проект — это как раз пример международного проекта третьего поколения, 100 тысяч азиатских геномов (GenomeAsia 100K). По словам докладчика, проект реализуется уже давно, и «довольно редко, но метко стреляет хорошими статьями». Первая из них, опубликованная в 2019 году в Nature, конструировалась в основном на населении Южной и Юго-Восточной Азии, Индии, Малайзии, Индонезии. Вторая — совсем свежая — вышла 15 мая в Science, и акцент в ней сделан на геномы населения Северной Евразии, Северной Азии и Южной Америки. (Подробнее в воскресном обзоре PCR.NEWS.) Называется она «From North Asia to South America: Tracing the longest human migration through genomic sequencing». Собранный в этой работе датасет содержит около полутора тысяч полных геномов, представляющих 130 популяций из Западной Евразии, Сибири, Северо-Восточной Азии, Южной Америки и ряда других регионов для референса. Вадим Степанов подчеркивает ценность этой информации — скажем, в этих 1,5 тысячах геномов обнаружили более 52 миллионов SNP и более 26 тысяч коротких инделов. Доля новых вариантов, описанных в этом проекте, очень велика — 60% обнаруженных SNP и около 90% инделов не содержались в существующих базах данных. Кроме того, показано, что разнообразие популяции человека с точки зрения генетической вариабельности очень сильно зависит от региона. Максимальное, как и следовало ожидать, наблюдается в Африке — в среднем геноме там насчитывается 4 миллиона полиморфных вариантов. Основная часть остального мира характеризуется уровнем 3–3,5 млн SNP на геном, а меньше всего их в Южной Америке, Сибири и некоторых небольших изолированных популяциях — индивидуальный геном может содержать даже менее 3 млн SNP.
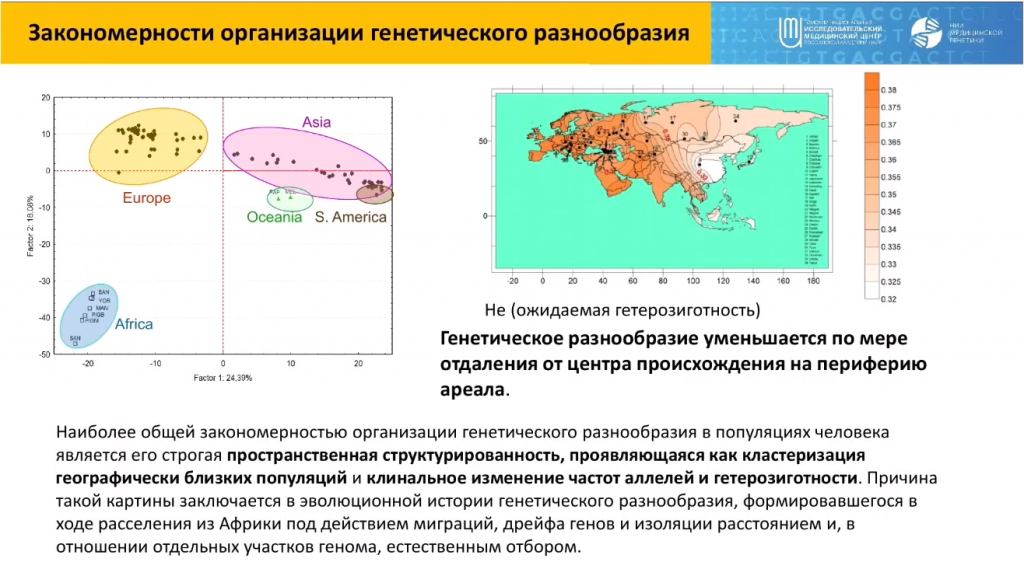
Как это разнообразие организовано в населении мира? Оно уменьшается по мере удаления человека от места происхождения. В результате популяции кластеризуются на геномной карте в первую очередь по географическим признакам, отвечающим продвижению человека в ходе расселения по земному шару.
И это разнообразие касается всего генома, в том числе и тех вариантов, которые так или иначе связаны с клиническими фенотипами. К примеру, разнообразие HLA аллелей — 18 у народов йоруба, живущих в Западной Африке, порядка 15 в Британии и всего около 5 в Восточной Сибири. Чем дальше человек расселялся по территории земного шара, тем большим количеством актов потери разнообразия это сопровождалось — так себя проявляет эффект бутылочного горлышка и дрейфа генов, которые, в частности, приводят к накоплению гомозиготных вариантов.
Очень показательным подходом для анализа генофондов популяций на уровне полных геномов является компонентный анализ, для которого статистически выбирают некое количество условных предковых компонентов, характерных для того или иного массива данных. В опубликованном массиве данных, который фактически охватывает весь мир, таких компонентов оказалось 18. В некоторых небольших изолированных популяциях встречаются уникальные компоненты — эти популяции особенно сильно отличаются от всего остального населения мира.
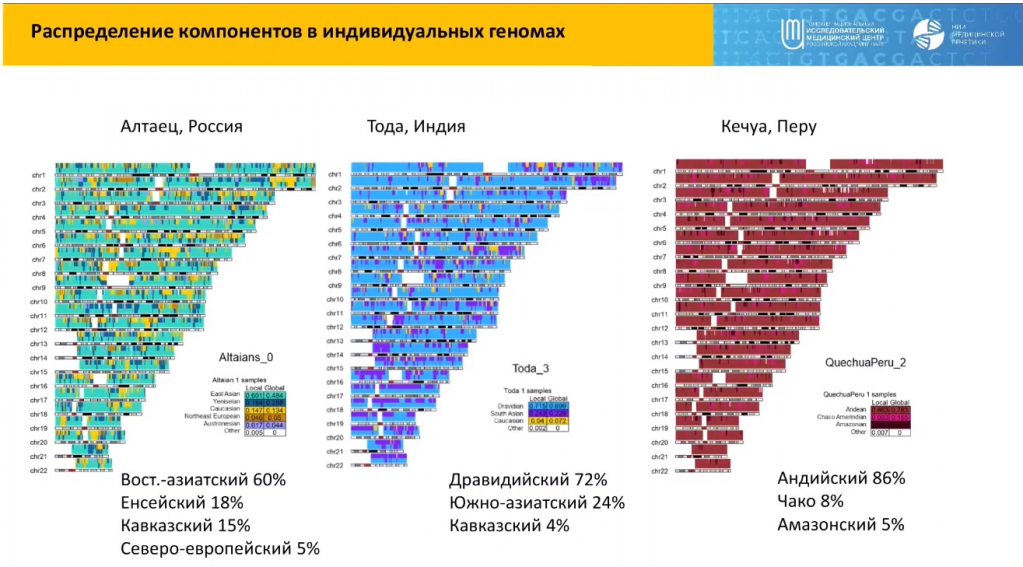
Каждый индивидуальный геном, подчеркивает докладчик, — это в некоторой степени смесь определенного количества предковых компонентов. В качестве примера он приводит геномы конкретных людей: алтайца из России, тода из Южной Индии и представителя племени кечуа из Перу.
Каждый из компонентов — это кусочек, унаследованный от какой-то из условных предковых популяций. У алтайца таким главным компонентом является восточноазиатский, у тода, как представителя дравидийской популяции из Индии — дравидийский, у кечуа из Перу — андский, и в целом у него представлены три из четырех южноамериканских компонентов.
Такого же рода логику — поиск участков генома и маркеров, специфичных для той или иной популяции, — использовали и в проекте по ДНК-идентификации. Анализ генома или некой выборки маркеров с него должен позволить предсказать или определить этнотерриториальное происхождение индивида только по его генетическим данным. Этот подход был реализован на выборке из примерно двух миллионов маркеров, из которых в итоговый вариант системы идентификации вошли около двух тысяч.
Современные геномные данные и подходы биоинформатики позволяют моделировать не только происхождение территориального генома или генофонда, но и анализировать древние изменения демографии популяции, то есть оценивать эффективную численность популяций в ходе тех или иных ключевых ветвлений. К примеру, разделение линий, ведущих к современным европейским популяциям Евразии и сибирским популяциям, произошло, по полученным оценкам, примерно 26–37 тысяч лет назад. Степень вариабельности высока, но эти данные совпадают со временем появления человека на территории Северной Евразии.
Следующее ключевое ветвление в районе 20–26 тысяч лет назад — это разделение предков современных североазиатских популяций и популяции американских индейцев. Важно, что модели также демонстрируют, как менялась эффективная численность популяции. Например, при разделении населения Северной Азии и Южной Америки она составляла примерно полторы тысячи человек. Затем, в силу разных особенностей генетики и демографических факторов, у кого-то численность росла, у кого-то уменьшалась. Южноамериканские индейцы сейчас — это представители народов или популяций с низкой численностью. Некоторые их популяции не насчитывают и сотни человек. Они находятся на грани вымирания — не только генетически, но еще и социально.
Что касается генетического груза, который несет каждый индивидуальный геном, то, разумеется, у каждого из нас есть масса неблагоприятных вариантов, связанных с моногенными болезнями, с побочным действием лекарств, с многофакторными заболеваниями. Однако распределение таких вариантов и сопоставление их с базами данных клинически значимых вариантов (например, ClinVar) показывает, что 99% геномов несут как минимум один патогенный вариант, каждый шестой — в гомозиготе. И далеко не у всех этих людей развивается заболевание. Объяснений может быть много — например, неполная пенетрантность или поздний возраст манифестации — но в некоторых популяциях они связаны с адаптивной значимостью вариантов, которые в других популяциях были бы патогенными.
Все это подчеркивает важность составления разнообразной выборке для полногеномных поисков ассоциаций — многие GWAS-исследования проводились на европейской популяции, упуская из внимания генетическое разнообразие многих других. Поэтому каждый такой проект, сделанный не в Европе, очень важен, в том числе и для клинической интерпретации вариантов, обнаруженных у тех или иных пациентов.
Пример неравномерного распределения вариантов, приведенный в докладе Вадима Степанова — хорошо известный вариант в гене бета-гемоглобина, связанный с устойчивостью к малярийному плазмодию. Адаптивность, а следовательно, и высокую распространенность он имеет только в африканских популяциях.
Еще один пример — ген карнитин-пальмитоилтрансферазы 1, зафиксированные в базе ClinVar патогенные варианты которого встречаются в некоторых популяциях Северо-Восточной Азии (в частности, у инуитов и эскимосов) более чем в 50% случаев. «Не может в популяции быть 50% носителей патогенного варианта. Эта мутация явно имеет для них адаптивную значимость», — говорит докладчик. Для доказательства этого можно провести тесты на естественный отбор — поиск сигналов гомозиготности указывает на накопление определенного варианта, анализ функциональной роли которого может указать на эволюционный смысл. Например, полученный сигнал в локусе 11q.13 (ген CPT1A) пришелся на патогенную мутацию, однако еt накопление в популяции связано с особенностями диеты северных народов — отсутствием растительной пищи и усиленным потреблением пищи животного происхождения.
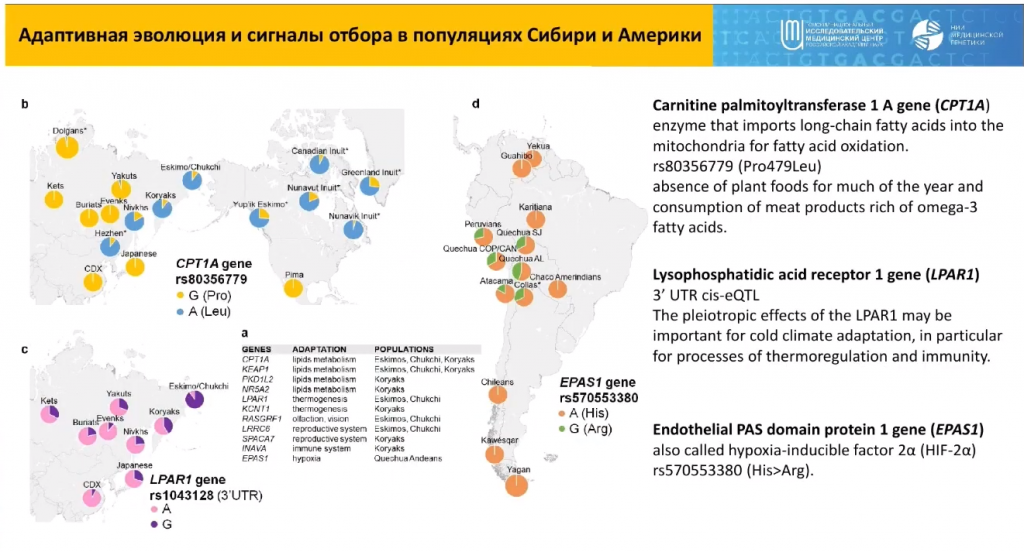
Из популяций Сибири и Америки наиболее интересными и генетически отличными от других являются популяции западной Берингии — те самые эскимосы, чукчи, коряки, — которые адаптировались к условиям Арктики путем накопления генетических вариантов, связанных с липидным обменом. Кроме того, их геномы обогащены сигналами отбора термогенеза, сенсорного восприятия и регуляции репродуктивных и иммунных функций.
«Популяционная геномика — замечательная наука, которая позволяет проводить высокоразрешающий анализ нашей генетической истории», — подытоживает докладчик.
Игорь Лебедев: «Полностью заменить цитогенетический анализ методами секвенирования не получается»
Доклад, посвященный цитогенетике, прочитал Игорь Лебедев, д.б.н., врио НИИ медицинской генетики и замдиректора по научной работе Томского НИМЦ.
Цитогенетика не так давно пережила переломный момент — в медицинскую генетику начали внедрять секвенирование генома человека и геномный анализ. Эти технологии можно применять и для диагностики хромосомных заболеваний. Именно эта возможность заставила некоторых специалистов задуматься: будут ли необходимы классические молекулярные тесты цитогенетики в эпоху секвенирования? В цитогенетическом сообществе возникли пессимистичные настроения; один из докладов, прочитанных британским генетиком Малкольмом Фергюсоном-Смитом на Европейской цитогеномной конференции, был озаглавлен «Хромосомное секвенирование, пятая и последняя эра цитогенетики».
Вопрос об актуальности цитогенетики действительно был дискуссионным, подчеркивает докладчик. Однако ответ на него приходит в наши дни, когда классические технологии во многом остаются востребованными.
На момент прочтения упомянутого доклада конференция уже называлась цитогеномной. Однако что вкладывается в этот неологизм — является ли «цитогеномика» простым объединением терминов «цитогенетика» и «геномика»? Является ли отрасль просто техническим объединением возможностей разных технологий в изучении хромосомного дисбаланса? Или же это объединение обладает каким-то синергичным эффектом, дающим новое знание о хромосомных заболеваниях?
Несмотря на масштабную трансформацию — на смену световой микроскопии приходит секвенирование хромосом, их исследования дополняются технологиями автоматизации и базами данных клинической значимости, — полностью заменить цитогенетический анализ методами секвенирования не получается. Они не дают представления ни о структуре хромосомной перестройки, ни о локализации перестроенных сегментов, полиплоидии, мозаицизме низкого уровня. Поэтому для детальной и полной характеристики хромосомного дисбаланса по-прежнему необходимы цитогенетические и молекулярно-цитогенетические технологии.
В качестве примера Игорь Лебедев приводит сравнение двух подходов к анализу хромосомных аномалий — однозначно выявить, что из-за перестройки возникла кольцевая хромосома, позволил только FISH, молекулярно-цитогенетический метод. Микроматричный анализ же не дает достаточно информации о кариотипе и не подтверждает наличие кольцевой хромосомы напрямую. Между тем выявление такой хромосомной перестройки совершенно меняет тактику медико-генетического консультирования семьи и возможность профилактики рождения больного ребенка через пренатальную диагностику.
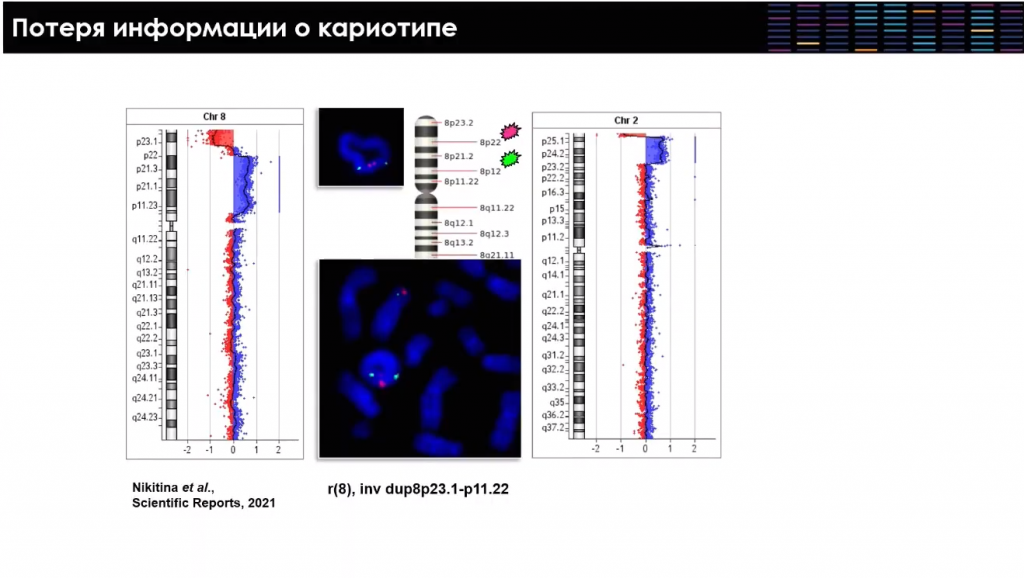
Некоторые типы хромосомного дисбаланса, в частности хромосомные микроделеции и микродупликации, связаны с неравным кроссинговером между блоками сегментных дупликаций. В последней версии сборки генома человека до 7% нашего генома представлено такими блоками сегментных дупликаций — их более 40 тысяч. Число выявленных микроделеционных синдромов растет, а также начинает увеличиваться количество известных микродупликационных синдромов, диагностика которых с использованием таргетных методов классической молекулярной цитогенетики оказывалась невозможной.
В настоящее время идет активное накопление информации о микроделеционных и микродупликационных синдромах — «болезнях-сестрах», как их называет идеолог этого направления Джеймс Лупски. Интересно, что классические микроделеционные синдромы, хорошо известные врачам-генетикам и цитогенетикам, совпадают по размеру и координатам перестроек с микродупликациями. Но каждый из «парных» синдромов — самостоятельное заболевание со своей собственной клинической картиной. Однако идентификация таких хромосомных изменений упрощается тем, что геномные технологии углубили понимание организации человеческого генома. Теперь, столкнувшись с неизвестными хромосомными дисбалансами, исследователь может сопоставить их с уже известными данными о хромосомных синдромах в этом регионе, полярными изменениями в числе копийности и прочей информацией. Иными словами, можно двигаться не только от клинической гипотезы и фенотипа к генотипу, но и в обратном направлении.
Примером из практики может служить пациент с микротрипликацией на длинном плече четвертой хромосомы. Этот вариант не был описан в литературе, а оценка патогенетической значимости подобных перестроек очень сложна. Связать эту перестройку с рядом дизморфических признаков и нарушением интеллектуального развития пациента было бы затруднительно, однако почти параллельно с этим в том же регионе была описана микроделеция, в которую попали ровно те же гены, что были затронуты исследуемой микротрипликацией.
Развивающийся организм может очень по-разному реагировать на полярные изменения в числе копий хромосомных регионов. Однако некоторые клинические признаки оказываются на удивление общими у пациентов как с отсутствием хромосомного участка, так и с увеличением его копийности. Обобщив, можно сказать, что микроделеционные и микродупликационные синдромы могут обладать общими, зеркальными и уникальными фенотипами.
Предложены три модели — аддитивная, доминантная и U-образная — которые объясняют появление того или иного фенотипического признака в зависимости от числа копий хромосомного региона.
Есть уже устоявшиеся классические примеры микроделеционных и дупликационных синдромов. Один из них – это реципрокные микроделеции на длинном плече первой хромосомы 1q21.1, которые ассоциированы с развитием микроцефалии, а реципрокная дупликация в этом же регионе приводит к макроцефалии.
Механизм такого проявления изучен — ген, затронутый перестройкой, определяет интенсивность движения ресничек, выстилающих желудочки головного мозга. Скорость движения целомической жидкости, с одной стороны, определяет параметры растущего мозга, а с другой, напрямую коррелирует с числом копий этого гена. Подобные обобщения появляются для многих других микроделеционных и дупликационных синдромов.
Однако картина генофенотипических корреляций может быть гораздо более сложной, и те общие признаки, которые мы видим у пациентов с делеционным и дупликационным вариантом, могут на самом деле встречаться с разной частотой.
Например, при перестройках терминальной области длинного плеча хромосомы 3 умственная отсталость и задержка развития чаще встречаются в случае дупликаций, а задержка развития речи, аномалии зрения, зубов, врожденные пороки сердца, напротив, в большей степени характерны для пациентов с делециями.
Чтобы объяснить подобные эффекты, необходимы модельные системы, обеспечивающие возможность изучения экспрессии генов и белков при таких хромосомных аномалиях. На протяжении нескольких последних лет исследовательская группа Игоря Лебедева вместе с коллегами из Института цитологии и генетики ФНКЦ физико-химической медицины ФМБА России проводит работы с ИПСК, полученными от пациентов с теми или иными патологиями, в том числе впервые описанными.
Одна из первых работ в этом направлении была связана с изучением эффекта хромосомных микроделеций и микродупликаций, затрагивающих другую часть хромосомы 3 — терминальную область короткого плеча. В них попадает единственный ген контактин-6, который ранее не был описан в отношении пациентов с умственной отсталостью. ИПСК, полученные от пациента с дупликацией, дифференцировали в нейрональном направлении и обнаружили неожиданное уменьшение уровня экспрессии дуплицированного аллеля. Оно фактически приводило к гаплонедостаточности и воспроизводило фенотип пациентов с микроделециями. Дополнительный вклад вносил эпигенетический контекст — обнаружилось, что нейроны здоровых доноров имеют разный уровень экспрессии с отцовского и материнского аллелей этого гена.
Дальнейшие работы были направлены на создание технологии хромосомного редактирования, которая позволила бы элиминировать дополнительную появившуюся копию гена в ИПСК этого пациента. Восстановить нормальную копийность хромосомного набора удалось, но эпигенетическая перестройка таким механизмом редактирования не снималась — на экспрессию гена оно не повлияло. Иными словами, хромосомное редактирование без эпигенетических модификаций вряд ли может изменить ситуацию.
В целом наличие хромосомной аномалии у кого-то из супругов — довольно частое событие для микроделеционных и дупликационных синдромов. Это указывает на важность оценки пенетрантности хромосомных перестроек. Как правило, в одних и тех же регионах дупликации имеют меньшую пенетрантность по сравнению с делециями. Такие особенности унаследованных хромосомных перестроек очень важно учитывать при проведении пренатальной диагностики.
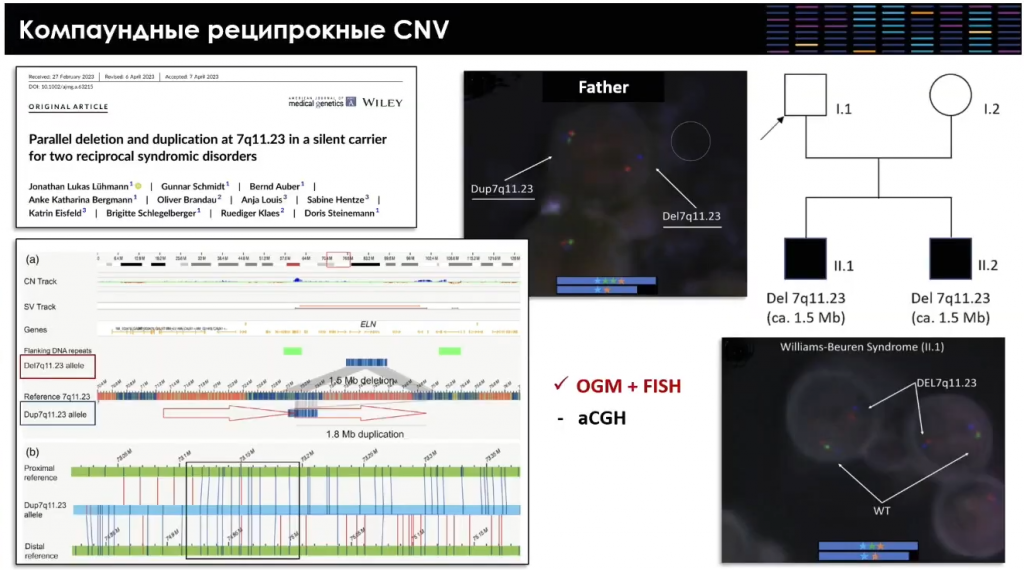
Докладчик рассказал об уникальном клиническом случае. В литературе описана семья, где два ребенка имели синдром Вильямса–Бойрена — классическую делецию на длинном плече хромосомы 7. Кариотипы родителей на первый взгляд казались нормальными. Однако после оптического картирования генома и FISH-теста оказалось, что отец — носитель одновременно делеции и дупликации, которые скомпенсировали друг друга. Еще одним механизмом могут служить аномалии X-хромосомы у матери, которые маскируются дозовой компенсацией, однако могут проявиться у ребенка мужского пола.
В клинике важно также учитывать возможность самокоррекции кариотипа — работы на ИПСК показали, что в ряде случаев кольцевая хромосома может элиминироваться из клетки, приводя к моносомии. Оставшийся гомолог дуплицируется с восстановлением нормального хромосомного набора, но фактически имеет место однородительская дисомия. Если на этих хромосомах нет импринтированных генов, то ситуация в целом благоприятная.
Но в случае с восьмой хромосомой не так все просто — на ней имеется ген калиевых каналов, экспрессирующийся только с материнской хромосомы. Кольцо у пациента было как раз материнского происхождения, и его элиминация привела к потере экспрессии гена KCNK9, который известен в контексте недавно описанного импринтированного синдрома Бирка-Барела (Birk-Barel syndrome).
В заключение Игорь Лебедев отметил, что использование цитогеномных технологий смещает акценты в современной молекулярной диагностике хромосомных заболеваний, но при этом по-прежнему актуальны молекулярно-цитогенетические подходы для детальной характеристики хромосомного дисбаланса. Все это приближает нас к пониманию генофенотипических корреляций при хромосомных заболеваниях. И, наконец, очень важный итог последних нескольких лет: с появлением высокоточной диагностики хромосомного дисбаланса, его происхождения и установления возможных наследственных вариантов появляются реальные перспективы профилактики рождения детей с хромосомной патологией в семьях с высоким генетическим риском через методы пренатальной диагностики и преимплантационного генетического тестирования.
Информация о докладчиках
Вадим Анатольевич Степанов, доктор биологических наук, профессор, академик РАН, директор Томского научно-исследовательского медицинского центра (НИМЦ), руководитель лаборатории эволюционной генетики НИИ медицинской генетики.
Игорь Николаевич Лебедев, доктор биологических наук, профессор, врио директора НИИ медицинской генетики Томского НИМЦ, заместитель директора по научной работе Томского НИМЦ, руководитель лаборатории онтогенетики.





 0
0