Меню
Меню





 Все темы
Все темы
Филогенетический анализ коронавирусов: нужно больше геномов
Исследования геномов коронавируса достоверно подтвердили местную передачу инфекции в штате Вашингтон. Однако происхождение итальянских вирусов из Германии, а также существование двух линий вируса, более опасной и менее опасной, не подтверждено.
Кристиан Дростен, вирусолог из университетской больницы Charité в Берлине, 28 февраля опубликовал геномную последовательность образца SARS-CoV-2, полученного от немецкого пациента, который был инфицирован в Италии. Он отметил сходство между этим геномом и геномом вируса первого мюнхенского пациента, опубликованным в конце января: три общие мутации, которых не было в геномах вирусов, опубликованных китайскими исследователями. Дростен предположил, что мюнхенский вирус мог быть занесен в Италию и вызвать там вспышку, однако возможен и другой сценарий: вариант с тремя мутациями мог независимо попасть в обе европейские страны. У себя в Твиттере он подчеркнул, что трех SNP «недостаточно, чтобы объявлять о связи между Мюнхеном и Италией», поскольку секвенировано слишком мало образцов вируса, и кроме того, в итальянском образце было 5 SNP, которых не было в мюнхенском. (Группа Дростена была первой, опубликовавшей протокол детекции нового вируса с помощью ОТ-ПЦР. Кроме того, Кристиан Дростен был одним из первооткрывателей коронавируса атипичной пневмонии SARS.)
Несколько дней спустя Тревор Бедфорд из Исследовательского центра по изучению рака Фреда Хатчинсона тоже написал об этом в Твиттере. Это сообщение широко распространилось, в публикации на MIT Technology Review говорилось также о сходстве мюнхенского кластера с новыми случаями в Мексике, Финляндии, Шотландии и даже Бразилии.
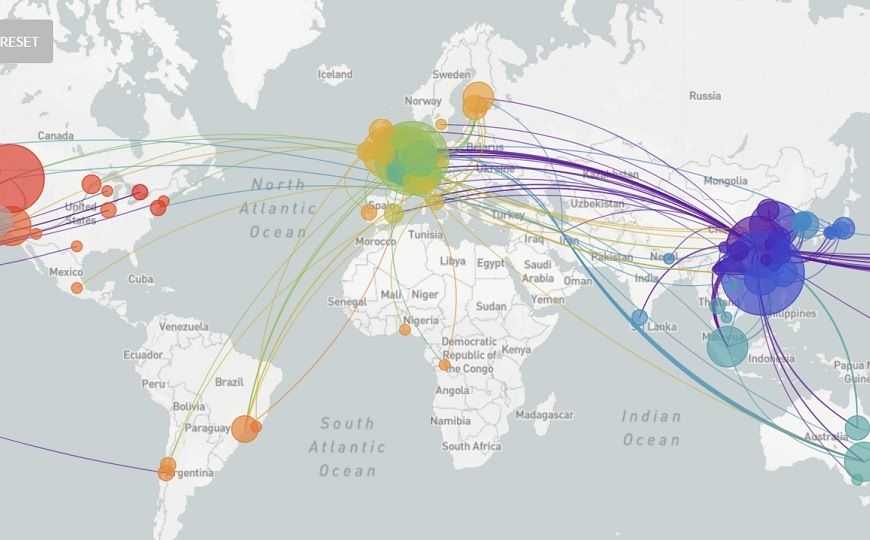
Однако и Кристиан Дростен, и другие специалисты по вирусной геномике сразу отметили, что данных для такого утверждения недостаточно и есть более вероятные сценарии попадания вируса в северную Италию, чем проникновение из Германии. Такую возможность допускал и сам Бедфорд, он добавил в Твиттере, что «следовало быть осторожнее» и что с появлением новых геномов картина может измениться. Действительно, уже сейчас эта часть филогенетического дерева на Nextstrain.org выглядит не так, как на скриншоте за начало марта.
Исследования геномов коронавируса дали достаточно много информации, в частности, подтвердили, что передача вируса от животного к человеку произошла единожды: если бы это происходило более одного раза, первые случаи заболевания у людей были бы генетически более разнообразными.
Теперь, по словам Эндрю Рэмбо, специалиста по молекулярной эволюции из Эдинбургского университета, в геноме SARS-CoV-2 длиной 30 000 нуклеотидов появляется в среднем от одной до двух мутаций в месяц, и это позволяет устанавливать связи между случаями.
Так, сходство между двумя геномами, секвенированными в штате Вашингтон, от пациентов, выявленных с разницей в шесть недель, указывало на то, что на протяжении этих шести недель вирус циркулировал в Вашингтоне незамеченным (подробнее на PCR.news). И Тревор Бедфорд, и другие эксперты согласились, что связь между этими двумя случаями намного более вероятна, чем независимое инфицирование первого и второго пациента похожими вирусами из Китая. К настоящему времени штат Вашингтон сообщил о более чем 160 случаях, и геномы от новых пациентов подтверждают первоначальное заключение.
Тем не менее даже полтысячи геномов (сейчас на Nextstrain.org представлено 519) — малая часть от всех случаев COVID-19, причем около 80% полученных последовательностей — из Китая. Вероятно, секвенирование новых геномов принесет больше информации.
Активно обсуждается возможность появления более или менее опасных линий коронавируса. Много споров вызвала работа, опубликованная 3 марта в китайском рецензируемом журнале National Science Review. Китайские исследователи проанализировали 103 генома и разделили их на две линии, отличающиеся двумя мутациями, которые назвали S и L, Поскольку 70% исследованных геномов SARS-CoV-2 принадлежали к более новому типу L, авторы сделали вывод, что этот тип эволюционировал, чтобы стать более агрессивным и распространяться быстрее.
Однако некоторые комментаторы считают эти выводы недостаточно обоснованными, китайцам даже советуют отозвать статью. «Одна из этих линий могла случайно оказаться больше другой», — говорит Эндрю Рэмбо. Критический разбор статьи опубликовали четверо ученых из Университета Глазго на сайте virological.org: «Заявления, сделанные в (статье), очевидно необоснованны и рискуют распространить опасную дезинформацию в критической ситуации вспышки». Ведущий автор, Лу Цзянь из Пекинского университета, ответил на это, что критика вызвана недопониманием.
Кристиан Дростен подчеркивает, что единственный надежный способ выяснить, насколько опасна мутация, — исследовать вирус в лаборатории и показать, что изменились характеристики его взаимодействия с клеткой.
Источник
Kai Kupferschmidt. // Genome analyses help track coronavirus' moves. // Science, 13 Mar 2020: Vol. 367, Issue 6483, pp. 1176-1177, DOI: 10.1126/science.367.6483.1176
Вам будет интересно

 2020
2020
 0
0


 1193
0
1193
0