
 Меню
Меню





 Все темы
Все темы
Новая мутация в гене CTNS у ребенка с цистинозом
У трехлетнего пациента с инфантильной нефропатической формой цистиноза российские генетики нашли ранее неизвестную протяженную делецию длиной 9 т.п.н., захватывающую регион от 3-го до 5-го интронов гена CTNS. Таким образом удалось поставить точку в «диагностической одиссее» ребенка.
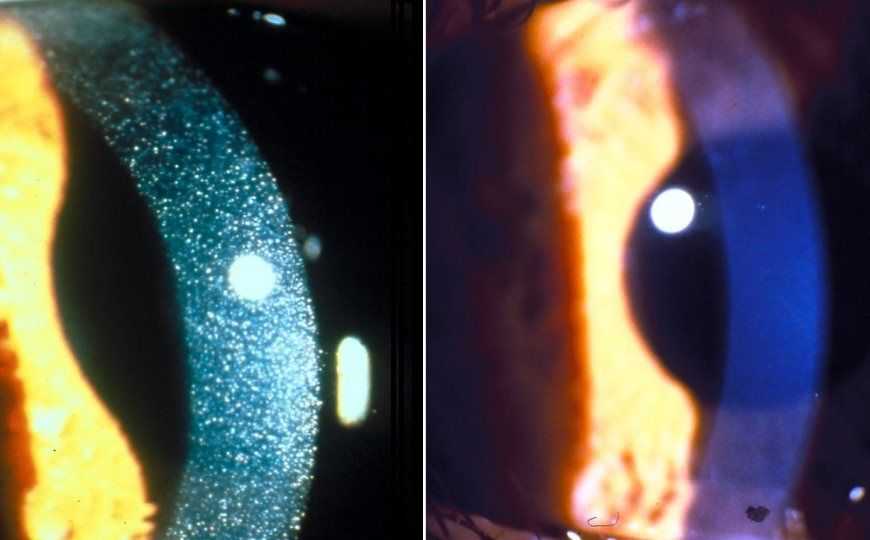
Фотография роговицы трехлетнего ребенка с нефропатическим цистинозом, сделанная с помощью щелевой лампы, до и после глазных капель с цистеамином. Слева хорошо видны кристаллы цистина. Credit: NIH Clinical Center Location: National Institutes of Health, Bethesda, MD - NHGRI genome.gov | Википедия
Работа выполнена в лаборатория функциональной геномики Медико-генетического научного центра (МГНЦ, Москва) совместно со специалистами по клинической генетики из НИКИ педиатрии им.Ю.Е. Вельтищева РНИМУ им.Н.И. Пирогова и сотрудниками Университетской больницы в Левене (Бельгия). Ребенка с подозрением на детский нефропатический цистиноз московские генетики начали наблюдать, когда ему исполнился год. Цистиноз — редкое аутосомно-рецессивное заболевание, при котором происходит внутрилизосомальное накопление цистина и отложение его кристаллов в различных органах и тканях. Выделяют три клинических формы болезни: наиболее тяжелая — классическая инфантильная нефропатическая форма цистиноза, неизбежно приводящая к терминальной уремии в первые десять лет жизни; ювенильная, или промежуточная форма с манифестацией в подростковом возрасте; и взрослая, «доброкачественная» форма цистиноза, проявляющаяся только поражением глаз с развитием фотофобии. Частота инфантильного нефропатического цистиноза в США и европейских странах составляет 1:100 000 – 1:200 000 живорожденных; ювенильная и взрослая формы отмечаются менее чем в 5% случаев.
Генетической причиной заболевания являются нарушения в гене CTNS, кодирующем лизосомальный переносчик цистина. Ген располагается на хромосоме 17p13 и состоит из 12 экзонов. Почти 75% всех исследованных на сегодня аллелей имеют делецию 57 т.п.н., затрагивающую область с 1-го по 10-й экзоны, хотя в базе HGMD описано не менее 120 других мутаций.
Развитие малыша в первые полгода соответствовало возрасту, однако в 7 месяцев родители заметили, что ребенок теряет вес и отстает в развитии, он стал больше пить, увеличился объем выделяемой мочи. К 12 месяцам у мальчика был выявлен полноценный синдром Фанкони, задержка роста, рахит и повреждения почек — медуллярный нефрокальциноз 1 степени. Однако при исследовании с помощью щелевой лампы в роговице глаза ребенка не было обнаружено кристаллов цистина — одного из главных клинических маркеров детского нефропатического цистиноза. Кроме того, исследование ДНК методом анализа полиморфизма длин рестрикционных фрагментов не выявило самой распространенной делеции в гене CTNS; секвенирование кодирующей части гена также не обнаружило отклонений в нуклеотидной последовательности. На некоторое время диагноз «нефропатический цистиноз» был снят для проведения дифференциальной диагностики с другими заболеваниями, при которых развивается синдром Фанкони: тирозинемия, галактоземия, гепаторенальный гликогеноз, болезнь Вильсона, синдром Лоу и т.д.
В возрасте 21 месяца повторное исследование выявило наличие кристаллов цистина в роговице, и клинический диагноз «нефропатический цистиноз» уже не вызывал сомнений; терапия цистеамином впоследствии улучшила состояние ребенка. Оставалось только подтвердить диагноз на молекулярном уровне. Поскольку проведенный ранее анализ гена не дал результатов, было принято решение поискать возможные причины заболевания глубоко в интронах или в регуляторных участках генома.
Чтобы проверить эту гипотезу, у ребенка взяли биопсию кожи, из этого образца получили первичную культуру фибробластов. Матричную РНК гена CTNS исследовали методом ПЦР с обратной транскрипцией, полученные фрагменты кДНК сравнивали с аналогичными фрагментами из фибробластов здорового донора. Секвенирование по Сэнгеру аномального (отличающегося по размеру) ПЦР-фрагмента обнаружило потерю 4-го и 5-го экзонов в мРНК пациента: мРНК теряет 159 нуклеотидов, что приводит к сдвигу рамки считывания и к появлению укороченной версии белка CTNS (p.Glu21GlyfsTer48).
При исследовании геномной ДНК пациента была обнаружена делеция размеров в 9 т.п.н., которая начинается в 3-м интроне гена CTNS и заканчивается в 5-м. Анализ ДНК матери и отца подтвердил, что оба родителя являются гетерозиготными носителями данной делеции (это неудивительно, учитывая кровное родство между ними). Таким образом, новый вариант аллеля NC_000017.10: g.3545967_3555253del был классифицирован как патогенный и занесен в базу данных LOVD (ID: 0000597339).
— Для нас всех этот случай был поучителен в том, что если врач убежден в поставленном диагнозе, то нужно постараться не только провести все возможные уместные анализы, но и в каких-то случаях и переделать анализ, начиная от самого начала — взятия нового образца от пациента, — говорит один из авторов исследования, зав. Лабораторией функциональной геномики МГНЦ Михаил Скоблов. — Дело в том, что сейчас мутации у больных с наследственными заболеваниями находят примерно в 30–40% случаев. И это пока абсолютно нормально, что для многих не находят, поскольку патогенные генетические варианты могут находиться глубоко в интронах или далеко в регуляторных участках, и поэтому искать их можно долго и это совсем не просто. Поэтому анализ РНК здесь очень информативен. При этом большинство наших пациентов приезжают издалека; вариант взять биопсию и получить из нее культуру клеток дает нам возможность проводить много разных анализов, проверяя различные гипотезы, поэтому Биобанк — это прекрасно (Биобанк — подразделение Центра коллективного пользования на базе ФГБНУ «МГНЦ» для систематизированного хранения поступающих образцов биоматериала. — PCR.news). Мы рады, что нам удалось закончить «диагностическую одиссею» для данного пациента и дать шанс семье планировать новую беременность и выносить здорового ребенка.
Источник
Papizh, S., et al. // CTNS mRNA molecular analysis revealed a novel mutation in a child with infantile nephropathic cystinosis: a case report. // BMC Nephrology, 2019, 20, 400; DOI: 10.1186/s12882-019-1589-2

Вам будет интересно


 116
116
 0
0










