Меню
Меню





 Все темы
Все темы
Геномы из России на эволюционном дереве SARS-CoV-2
Ученые всего мира получают геномные последовательности коронавируса SARS-CoV-2. На сегодня в глобальной базе данных более 34 тысяч геномов, из них 207 из России. Конечно, это немного, но гораздо лучше, чем ничего. Мы поговорили со специалистами, которые исследуют генетические и эволюционные особенности возбудителя пандемии COVID-19.
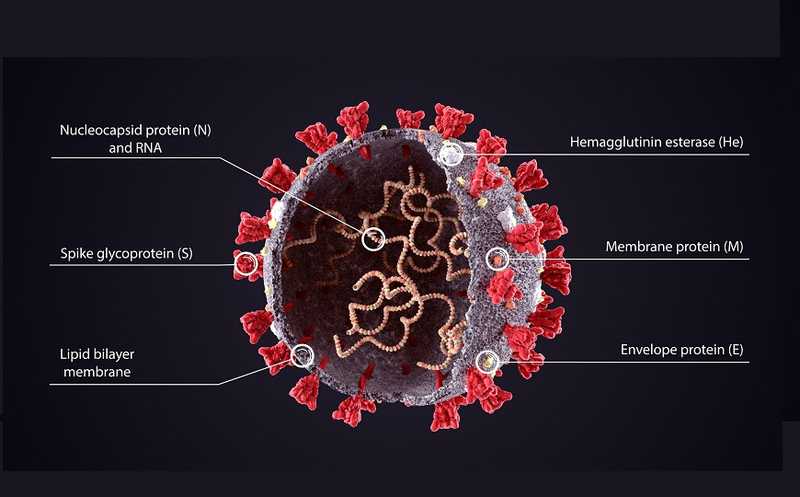
Молекулярные биологи секвенируют геномы SARS-CoV-2, полученные из проб пациентов в разных точках планеты, биоинформатики сравнивают нуклеотидные последовательности. Анализ возникающих в них мутаций служит для построения филогенетических деревьев, которые показывают родственные связи между разными штаммами вируса. Филогенетический анализ SARS-CoV-2 важен для нескольких практических задач. Выявление родственных связей между разными вариантами вируса позволяет предположительно реконструировать пути его передачи и проследить распространение. Вместе с тем секвенирование большого количества геномов необходимо, чтобы понять особенности эволюции нового вируса, скорость и распределение мутаций, а это очень ценная информация для создания вакцины.
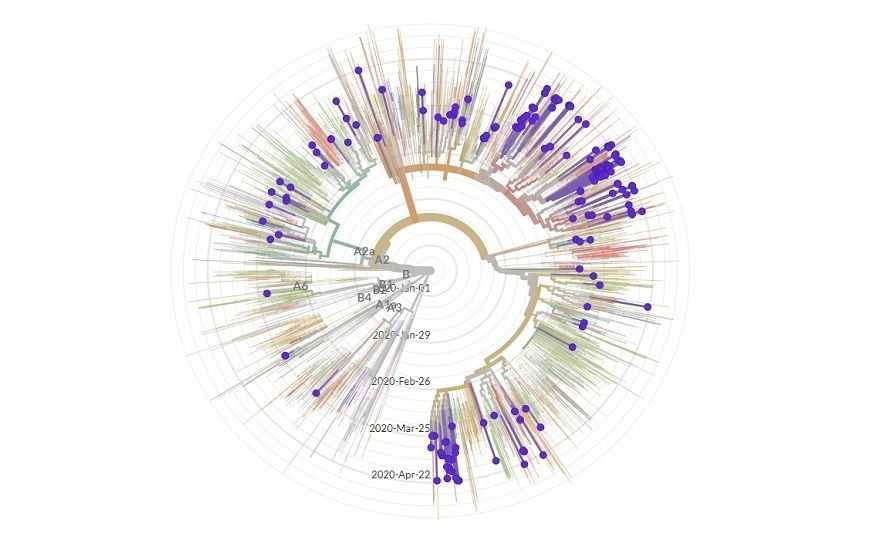
Наиболее популярная глобальная база вирусных геномов — GISAID, любой исследователь после регистрации может загрузить в нее данные своего сиквенса SARS-CoV-2. На текущий момент в этой базе более 34 тысяч геномов со всего мира, и она все время пополняется. Данные из разных стран включаются в филогенетический анализ: специальный алгоритм фильтрует последовательности по качеству, по длине, определяет нуклеотидные замены. Результаты анализа визуализируются на сайте Nextstrain в виде филогенетических деревьев и предположительной реконструкции путей передачи вируса на географической карте. Можно посмотреть общую картину в масштабе всего мира или региона, например, Европы. Можно отфильтровать данные по нашей стране: российские геномы коронавируса на ветвях дерева Nextstrain разбросаны яркими точками, их число увеличивается на глазах, сегодня там 202 генома. Уточним, что в базе GISAD на сегодня 207 геномов из России, но для филогенетического дерева отбирают не все.
Коронавирус мутирует относительно медленно
Первыми на дереве появились геномы из Санкт-Петербурга, их секвенируют и выкладывают в базу GISAD специалисты НИИ гриппа им. А.А.Смородинцева, на сегодня они выложили 136 сиквенсов. Сейчас география значительно расширилась: есть геномы из Москвы, Самары, Пскова, Краснодара, Екатеринбурга, Красноярска, Челябинска, Хабаровска, Якутии, Татарстана, Бурятии и других регионов. Большая их часть секвенирована специалистами ГНЦ вирусологии и биотехнологии «Вектор». Московские геномы поступают из НИЦ эпидемиологии и микробиологии имени Н.Ф. Гамалеи, ФНЦ исследований и разработки иммунобиологических препаратов им. М.П. Чумакова.
Мы поговорили с заведующим лабораторией молекулярной вирусологии в НИИ гриппа Андреем Комиссаровым, автором первых сиквенсов российских геномов на коронавирусном дереве. Его лаборатория проводит тестирование на COVID-19 для города. Кроме того, клинические образцы поступают из стационаров и поликлиник по проекту госпитального надзора над гриппом и ОРВИ, среди них выявляются и геномы коронавирусов, как сезонных, так и нового. Первый геном SARS-CoV-2 специалисты прочитали 15 марта, по словам Комиссарова, он оказался наиболее близок к штамму из Франции.
«Мы быстро включились в процесс сбора геномных данных по коронавирусу, потому что уже давно этим занимаемся по гриппу, — рассказывает Андрей Комиссаров, — мы секвенируем геномы вируса гриппа и выгружаем на тот же сайт. Nextsrain, он же не только про коронавирус, он исходно создавался как сайт Nextflu, был заточен на грипп. По числу проанализированных полных геномов вируса гриппа мы вышли на второе место в Европе после Великобритании. Когда началась история с коронавирусом, мы поняли, что рано или поздно он попадет в Россию, и нужно быть готовыми к генетическому анализу. Как только в марте образцы наконец-то попали к нам в руки, мы буквально за два дня секвенировали первый полный геном».
Какую информацию специалисты получают из прочитанных геномов коронавируса? Прежде всего, анализ числа и распределения мутаций показывает изменчивость вируса, скорость и закономерности его эволюции. «Нас интересует распределение нуклеотидных замен по разным генам, — объясняет Андрей Комиссаров. — Очень интересуют замены в сайтах связывания праймеров различных тест-систем. Анализ сайтов посадки праймеров нужен, чтобы понимать, насколько хорошо работают тест-системы и возникают ли варианты вируса, на которых тест-системы будут работать плохо».
Что касается скорости, то оказалось, что по сравнению с другими ОРВИ-вирусами и с вирусом гриппа SARS-CoV-2 эволюционирует довольно медленно. Скорость его мутирования оценили величиной 8 х10-4 на сайт в год (правда, она была подсчитана, когда геномов было значительно меньше). По словам Комиссарова, это на порядок меньше, чем у вируса гриппа. О небольшой скорости мутаций говорит и то, что первый секвенированный петербургский геном продемонстрировал пять несинонимичных замен (оказывающих влияние на синтез белка) относительно уханьского штамма, это немного. И это очень хорошо для создания вакцины.
Филогения с географией
Геном SARS-CoV-2 на дереве Nextstrain, поступивший из НИЦ эпидемиологии и микробиологии имени Гамалеи Минздрава России, секвенирован в референс-центре по коронавирусам. Как объяснил руководитель референс-центра Владимир Гущин, это геном вируса, который был выделен от пациента и размножен на культуре клеток, что обеспечило высокое качество сиквенса. Задача исследователей — не просто секвенировать, а пополнять государственную коллекцию вирусов, содержащую живые образцы, которые в любой момент могут быть использованы для проверки тест-систем, создания вакцин и разработки лекарственных препаратов. По словам Гущина, геном вируса, выделенного у московского пациента, оказался близок к штаммам, которые циркулируют в Европе и Северной Америке, но более всего похож на британские штаммы.
Вопросы распространения нового коронавируса по миру и источники его происхождения в каждой стране — одни из самых интригующих. На сайте Nextstrain имеется карта, на которой реконструированы пути передачи вируса. Эту карту можно запустить в анимированном режиме и наблюдать, как цветные линии протягиваются от одной страны до другой, постепенно связывая регионы мира наподобие рейсов авиакомпаний. Параллельно показана динамика ветвления дерева с предположительным временем возникновения новых ветвей. Если верить этой карте, инфекция появилась в России в конце февраля и завезена была из Италии. Но пока что число секвенированных геномов из России слишком мало по сравнению с данными из других стран.
«Любая филогеографическая и филодинамическая реконструкция требует большого массива данных. Тех данных, которые мы имеем сейчас в России, для этого недостаточно, — считает Андрей Комиссаров. — Сейчас самая главная задача — сделать так, чтобы данных из России было много. Очень важно, чтобы как можно больше лабораторий подключались к этому и секвенировали геномы».
С тем, что нужно больше геномов, согласен Георгий Базыкин, профессор Сколтеха, зав. лабораторией молекулярной эволюции Института проблем передачи информации РАН. Его группа занимается филогенетическим анализом геномов SARS-CoV-2 и сотрудничает со специалистами НИИ гриппа с тех пор, как они прочитали первый российский геном. «Для молекулярной филогенетики чем больше последовательностей, тем лучше, — говорит Базыкин. — Но здесь важно не только число. Эти последовательности приобретают силу, если их удается сопоставить с эпидемиологическими данными, если известен не только город, откуда они взялись, не только дата взятия образца, но и детали. Если связать эпидемиологические данные с молекулярными, то мы можем делать гораздо более интересные выводы». Полезные с точки зрения эпидемиологии данные включают маршрут передвижения пациента, если перед заболеванием он посещал какую-то страну или город внутри страны; люди, с которыми он контактировал, — заболевшие, от которых он мог заразиться, и здоровые, которые могли подхватить вирус от него.
Интересно разобраться в том, завозные те или иные варианты вируса или местные. Сначала, в марте, большинство случаев инфекции были завозными. Анализ первого секвенированного российского генома показал, что он расположился внутри большой европейской клады и эволюционно ближе всего к штамму, выделенному во Франции. Как прокомментировал Георгий Базыкин в своем блоге, это «сильное свидетельство в пользу завоза в Россию из Европы». Постепенно доля завозных случаев инфекции падала и все больше становилось случаев, связанных с местной передачей.
«Интерпретировать с осторожностью»
Насколько точна карта трансмиссии коронавируса на сайте Nextstrain? «В каких-то своих аспектах она точна, в каких-то аспектах совсем неточна, — считает Базыкин. — Создатели ресурса это многократно подчеркивают. Предположим, например, что вы видите две ветви, одну французскую, одну итальянскую, отходящие от одного корня. Был здесь перенос из Италии во Францию или наоборот? Глядя только на дерево, мы этого сказать наверняка не можем. Теперь предположим, что некоторая ветвь включает несколько последовательностей, выделенных у разных пациентов, и все эти случаи — в одном городе. Первая интерпретация, которая приходит в голову — что вирус передается внутри этого города. Но возможно и другое — что все мутации в этой кладе произошли еще до завоза в этот город, там, откуда вирус пришел. Просто у нас не было достаточного количества образцов из того места, откуда он пришел, и мы этого не распознали. Поэтому все результаты филогенетического анализа нужно интерпретировать с большой осторожностью. Особенно аккуратно нужно интерпретировать датировки, потому что это модельные оценки, и в этих моделях делается много предположений».
Как упомянул Комиссаров, первые случаи COVID-19 в России произошли в Тюмени и Забайкальском крае, но их возбудители не были генетически изучены. По мнению Базыкина, если бы те случаи породили вспышки заболевания, то мы бы об этом знали. Возможно, они были успешно изолированы и не распространились. Все же последующие кластеры передачи вируса в России в основном имеют европейское происхождение.
Знания о возбудителе пандемии COVID-19 добываются всем миром, каждая страна вносит вклад в глобальное исследование. Из США и западноевропейских стран поступают тысячи геномов. Количество данных из России пока несопоставимо меньше,. Очень жалко, что в базе мало московских геномов, так как Москва в эпидемиологической ситуации в нашей стране играет основную роль.
В целом, чем больше секвенированных геномов, тем полнее филогенетический анализ, тем точнее реконструкции. Чтобы увеличить российский вклад в глобальное исследование, специалисты НИИ гриппа в Санкт-Петербурге создают консорциум по секвенированию геномов SARS-CoV-2 и приглашают всех к сотрудничеству. Желающих уже много, и ученые надеются, что консорциум заработает в ближайшее время. Готовятся и научные публикации по филогении коронавируса, ждем их на сайте препринтов.

Вам будет интересно


 2360
2360
 0
0