Меню
Меню





 Все темы
Все темы
Генное редактирование клеток мозга смягчило симптомы альтернирующей гемиплегии у мышей
Альтернирующая гемиплегия детского возраста — редкое тяжелое заболевание с неврологическими симптомами, вызванное мутациями в гене субъединицы натрий-калиевой АТФазы. Генная терапия с доставкой гена в нейроны не привела к клиническому улучшению на мышиных моделях заболевания. Однако праймированное редактирование в клетках мозга in vivo смягчило симптомы у мышей и увеличило продолжительность их жизни.

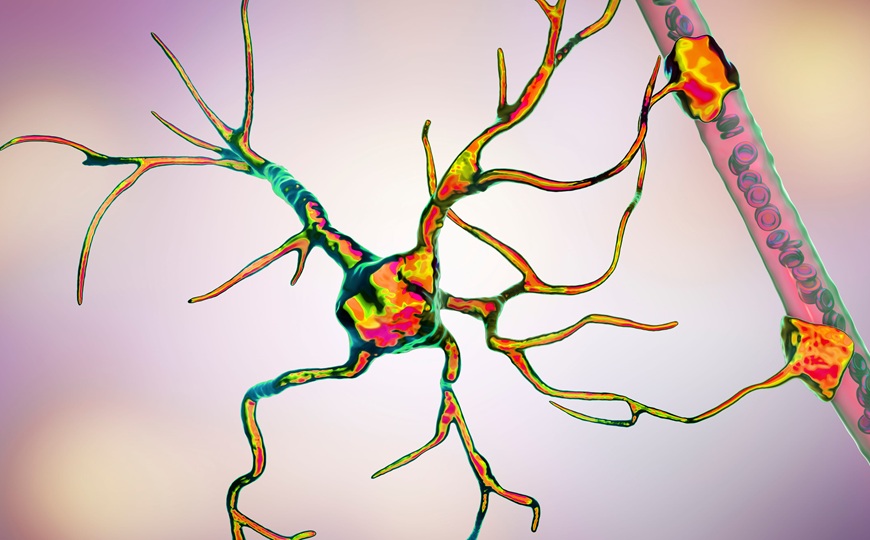
Судороги у мыши, вызванные охлаждением.
Credit:
Cell. 2025. DOI: 10.1016/j.cell.2025.06.038 | CC BY 4.0
Все больше препаратов для генной терапии получает разрешение регуляторов. Есть сообщения об успехах генного редактирования in vivo (в организме пациента). Однако головной мозг — сложная мишень. Редактирующие конструкции в липидных наночастицах легко доставить, например, в клетки печени (именно так сделали с индивидуально разработанным CRISPR-редактором для маленького ребенка). Однако мозг защищен гематоэнцефалическим барьером, и ввести в его клетки генотерапевтическую или редактирующую конструкцию намного сложнее.
Исследователи из США и Франции представили результаты эксперимента по редактированию in vivo мутаций, которые вызывают альтернирующую гемиплегию детского возраста. Симптомы этого редкого наследственного заболевания (частота около 1 случая на 1 млн человек) проявляются до 18-месячного возраста. Эпизоды гемиплегии, то есть потери подвижности ноги и руки с одной стороны тела, могут продолжаться от нескольких минут до нескольких дней, возможно развитие других неврологических симптомов, включая судороги и когнитивные нарушения.
До 70% случаев заболевания вызывают мутации в гене ATP1A3, кодирующем субъединицу Na+/K+ АТФазы. В исследовании на мышиной модели, где в клетки доставлялся ген без мутации, к сожалению, не наблюдалось клинически значимых улучшений.
«Это ужасная болезнь», — говорит Дэвид Лю из Института Брода Массачусетского технологического института и Гарвардского университета, один из создателей первых редакторов оснований. (Мы недавно писали о применении этого подхода для терапии серповидноклеточной анемии; в этом случае редактирование клеток проводилось вне организма.)
Для редактирования мышиных моделей альтернирующей гемиплегии Лю и его коллеги применили два метода — редактирование оснований (base editing, BE) и праймированное редактирование (прайм-редактирование, prime editing, PE). При редактировании оснований происходит замена единичных нуклеотидов, а с помощью прайм-редактирования можно заменять протяженные участки генома. Но в обоих случаях не делаются двунитевые разрывы в ДНК и не нужна внесенная извне ДНК-матрица с желаемой последовательностью, что снижает вероятность нецелевых изменений, таких как делеции и инсерции.
Уже проводится клиническое исследование терапевтического РЕ ex vivo хронической гранулематозной болезни, но в организме, тем более в клетках мозга, подобные подходы еще не применялись.
Авторы исправляли мутации, связанные с альтернирующей гемиплегией детского возраста, в иПСК, полученных от пациентов, с помощью ВЕ и РЕ. Мишенями редактирования были пять мутаций, которые в совокупности вызывают более 65% связанных с ATP1A3 случаев заболевания. В иПСК удалось исправить 40–90% мутаций, значительно снизив при этом уровень нецелевого редактирования, особенно в экспериментах с РЕ.
Важно было проверить, смягчает ли редактирование патологию альтернирующей гемиплегии. Для этого использовали две линии мышей с двумя мутациями, соответствующими двум самым распространенным патогенным вариантам ATP1A3 у человека. Новорожденным мышатам вводили конструкции для прайм-редактирования в векторах на основе аденоассоциированного вируса AAV9 (PE-AAV); инъекции делались в желудочки мозга. Доза вектора была ниже, чем у широкоизвестного препарата Золгенсма, который тоже содержит AAV9.
Мутации удалось исправить в значительной доле клеток коры головного мозга и мозжечка. У взрослых мышей наблюдались улучшения по ряду показателей. Пароксизмальные реакции и судороги, вызванные охлаждением, стали менее выраженными, улучшились когнитивные функции и контроль движений, а продолжительность жизни увеличилась. Например, среди самцов и самок мышей с мутацией D801N, получавших редактор, 56% и 77% соответственно дожили до 52 недель, тогда как все самцы с такой мутацией, получившие плацебо, умерли до 35-й недели, все самки — до 51-й, со средним временем выживания 13 и 25 недель соответственно. «Мы были поражены», — сказал Дэвид Лю.
Лаборатория Лю также исправила у мышиных моделей мутации, вызывающие два других неврологических расстройства: болезнь Хантингтона и атаксию Фридрейха (подробнее на PCR.NEWS). А в Медицинской школе Шанхайского университета Цзяотун в Китае нейробиолог Цзылун Цю и его коллеги с помощью редактора оснований использовали редактирование оснований для коррекции мутации в гене Mef2c у мышей. У детей мутация в соответствующем гене может вызывать эпилепсию, умственную отсталость и задержку речевого развития.
Цю и Лю независимо друг от друга работают над методами генной терапии синдрома Ретта, который чаще всего вызывается мутациями в гене MECP2. В данном случае генное редактирование предпочтительнее внесения в клетку активной копии гена, поскольку избыток его продукта может быть токсичным.
Обе команды предполагают, что в клинических испытаниях для доставки в мозг будут использоваться производные аденоассоциированного вируса 9 (AAV9), который способен проникать через гематоэнцефалический барьер и инфицировать клетки мозга. Однако высокие дозы AAV9 могут спровоцировать жизнеугрожающую иммунную реакцию. Исследователи продолжают разрабатывать более безопасные вирусные векторы, которые можно использовать в низких дозах.
Первое в мире индивидуальное генетическое редактирование в организме пациента прошло успешно
Источник
Alexander A. Sousa, et al. In vivo prime editing rescues alternating hemiplegia of childhood in mice // Cell. Volume 188, Issue 16P4275-4294.E23 August 07, 2025. DOI: 10.1016/j.cell.2025.06.038

Вам будет интересно

 57
57
 0
0


 52
0
52
0