
 Меню
Меню





 Все темы
Все темы
Виктор Татарский: «Все, что я вам рассказал, — это немножко ложь или сильное упрощение»
Новый лекционный блок научно-просветительского проекта «Метафаза» от PCR.NEWS и парка «Зарядье» продолжил Виктор Татарский. Он рассказал о многочисленных белках, регулирующих клеточный цикл, и о нарушениях, приводящих к опухолеобразованию. А также о том, почему неделящиеся раковые клетки — это плохо, как клеточное старение провоцирует развитие рака, можно ли убить опухолевую клетку, заставив ее делиться еще быстрее, и почему так сложно воспользоваться знаниями о клеточном цикле для разработки лекарств.

Виктор Татарский, кандидат биологических наук, заведующий лабораторией молекулярной онкобиологии Института биологии гена РАН, выступил с лекцией «Регуляция клеточного цикла и его нарушения в канцерогенезе»
Клеточный цикл в целом
Клеточный цикл делят на две большие части: интерфазу и митоз. Интерфаза, в свою очередь, подразделяется на три фазы: G1, или постмитотическая пресинтетическая фаза, S — синтетическая фаза, или фаза репликации, и G2, или постсинтетическая премитотическая фаза. Но на самом деле многие клетки организма находятся в так называемой фазе G0, она же фаза покоя (quiescent cell). Это клетки, которые не получили сигнала о том, что им нужно поделиться. Соответственно, они не делятся, но ожидают соответствующего сигнала. Помимо этого, есть терминально дифференцированные клетки, которые в принципе уже не могут делиться. Это, например, нейроны или кардиомиоциты.
Существует еще одно интересное состояние, так называемое клеточное старение. По-английски это cellular senescence, поэтому также эти клетки называют сенесцентными. Это клетки, которые были повреждены или достигли предельного количества делений, и дальше они делиться уже не могут. Хотя в опухолях бывает так, что такие клетки выходят из этого состояния.
В норме, чтобы клетка начала делиться, она должна получить соответствующий сигнал, при этом у нее должны быть все ресурсы для деления, а никаких сигналов о том, что ей нельзя делиться, она не получала. Соответственно, решение о том, входить или не входить в клеточный цикл, зависит от многих белков, которые следят за состоянием клетки. Даже если клетка получила сигнал о том, что ей нужно поделиться, но при этом ей не хватает каких-то ресурсов, она не прикреплена к субстрату, или пришли сигналы, которые говорят о том, что ей нельзя делиться, например, потому что ее ДНК повреждена, — она делиться не будет.
Существует множество сигналов, которые могут побудить клетку делиться, но в любом случае при этом начинает экспрессироваться циклин D1, белок, который нужен для вхождения в клеточный цикл.
Циклины и циклин-зависимые киназы
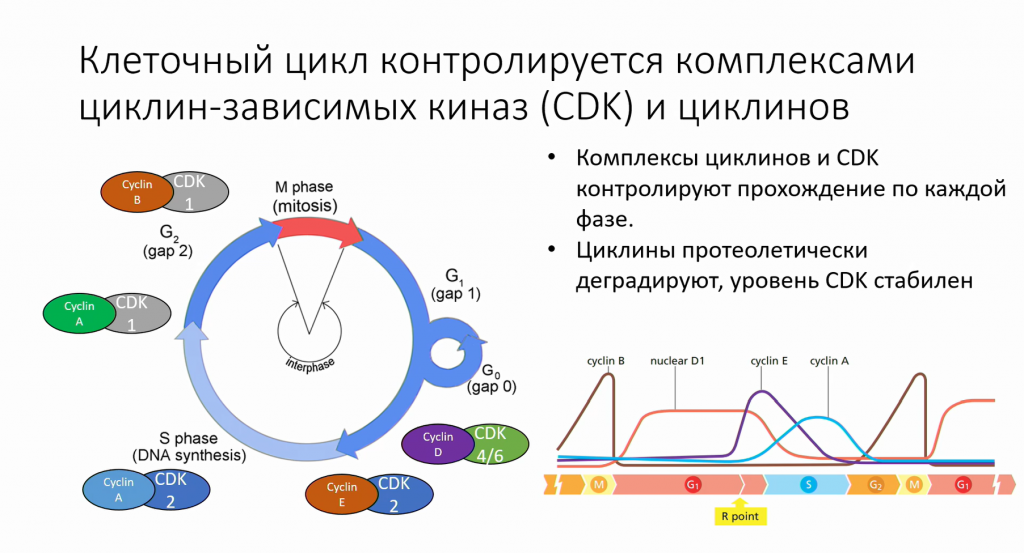
Циклины и циклин-зависимые киназы (CDK) — это основные белки, которые управляют прохождением по клеточному циклу из одной фазы в другую. Киназы — ферменты, которые переносят фосфаты на свои субстраты, их число входят протеинкиназы — киназы, которые переносят фосфаты на другие белки. Обычно при этом меняется конформация белков, и это приводит к их активации или деактивации, к изменению стабильности и т. д. Циклин-зависимые киназы могут работать только вместе со своими циклинами. Обычно уровень киназ довольно стабилен, а вот циклины появляются в клетке только в той фазе, которую они контролируют, а потом быстро деградируют. Циклин D1 здесь является исключением, потому что он не деградирует, но уходит из ядра.
Циклин Е и циклин-зависимые киназы 2 контролируют вход в S-фазу, затем циклин Е деградируют, его заменяет циклин А, он контролирует начало и окончание репликации. Затем он деградирует и заменяется циклином B, а циклин-зависимая киназа 2 в процессе заменяются на циклин-зависимую киназу 1. Потом происходит резкая деградация циклина B, после разделения клеток в митозе. В начале каждой фазы циклины начинают экспрессироваться и стабилизироваться, и в конце каждой фазы они прекращают транскрибироваться и одновременно помечаются для удаления. В норме их уровень падает быстро.
Контрольные точки
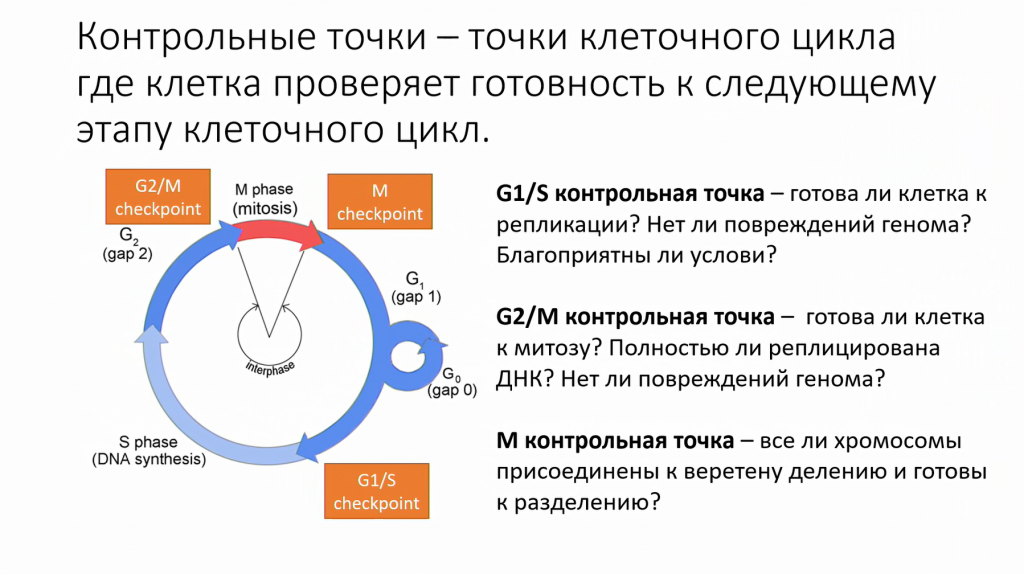
Клетка не просто двигается по этим фазам цикла, она очень осторожно относится к тому, чтобы не входить в необратимое состояние деления, если она к этому не готова. Здесь есть так называемые контрольные (сверочные) точки, или чекпоинты, в которых клетка проверяет, готова ли она к следующему этапу.
В G1/S контрольной точке клетка проверяет, что у нее есть все необходимые белки для репликации, что они сидят в ориджинах репликации на ДНК, что нет значительных повреждений ДНК и нет мутаций, которые могут превратить клетку в опухолевую.
Если клетка, например, имеет плотные клеточные контакты с другими клетками, она просто так тоже делиться не будет. Есть разные отрицательные сигналы, которые не дадут ей продвинуться вперед.
Затем в G2/M контрольной точке клетка проверяет, что вся ДНК была реплицирована и при этом нет повреждения генома, и что у нее есть все необходимые белки для митоза.
Клетка вступает в митоз, хромосомы выстраиваются в митотическом веретене, и здесь есть последний чекпоинт, так называемый митотический чекпоинт, когда клетка проверяет, что все хромосомы правильно выстроены и прикреплены к веретену деления и что сила натяжения веретена одинаковая, так что они правильно разделятся. Если все эти условия соблюдены, то произойдет митоз.
Уровни контроля
Контрольные точки и в целом активность циклин-зависимых киназ регулируются сразу на нескольких уровнях. Это уровни транскрипции, белковой стабильности и посттрансляционных модификаций. Все они влияют на то, есть ли в клетке активные комплексы циклин-зависимых киназ и циклинов.
Собранные комплексы циклин-зависимых киназ и циклинов должны быть правильно фосфорилированы. Есть активирующее фосфорилирование, которое добавляет так называемая CAK (CDK активирующая киназа). Ингибирующий фосфат добавляют киназы семейства Wee1 и удаляют фосфатазы CDC25.
Следующий уровень, который регулирует активность комплексов циклин-зависимых киназ и циклинов, — это белки-ингибиторы циклин-зависимых киназ, или CDK inhibitor proteins (CDKi). Это небольшие белки, которые физически связывают эти комплексы, блокируют их карман, который взаимодействует с лигандами, и не дают им фосфорилировать свои субстраты. Они делятся на две группы. Одни называются семейством INK4. Это белки p16, p15, p18, p19, которые могут связываться только с комплексами циклина D и CDK4/CDK6. Соответственно, они могут остановить клеточный цикл в G1. Второе семейство CDKi — Cip/Kip, это белки p21, p27, p57, которые могут останавливать все остальные комплексы CDK2 и CDK1 и блокировать циклы в других фазах.
Эти белки очень маленькие, они очень быстро могут транскрибироваться и затем так же быстро деградируют. Соответственно, клетка может быстро увеличить их количество, и они блокируют активность комплексов CDK-циклинов. Их транскрипция и стабильность увеличиваются при повреждении ДНК, активации ингибиторных сигнальных каскадов типа TGF-бета, гипоксии, отсутствия связи с внеклеточным матриксом, отсутствия влечения к митотическому веретену и так далее. Иначе говоря, при стрессе эти белки быстро накапливаются и блокируют активность комплексов CDK-циклинов.
Белки Cip/Kip блокируют комплекс CDK2 с циклином E, но, с другой стороны, стимулируют образование комплекса CDK4/6/циклин D1 и доставляет его из цитоплазмы в ядро. Их некоторое количество необходимо для того, чтобы инициировать переход в клеточный цикл из состояния покоя. И их ингибиторные и активирующие активности зависят от фосфорилирования. Например, ядерный p27 работает как опухолевый супрессор, не дает клетке делиться, а если он находится в цитоплазме, то работает больше как онкоген. У пациентов с p27 в цитоплазме хуже прогноз.
Следующий уровень регулирования — стабильность белка. Когда циклины перестают быть нужными, E3-лигазы навешивают на них полиубиквитиновые цепи, они отправляются в протеасому и деградируют.
С другой стороны, ингибиторы циклин-зависимых киназ, как только стресс прошел, например, клетка репарировала повреждения, тоже быстро помечаются на деградацию лигазами и отправляются в протеасому. E3-лигаз в клетке очень много, у них у всех свои субстраты. У субстратов клеточного цикла есть различные E3-лигазы, которые активны в разных фазах цикла. И их активность настолько точна, что используется для одного из довольно популярных методов наблюдения за циклом в живой человеческой клетке (окраска Фуччи — Fluorescent Ubiquitination-based Cell Cycle Indicator). В клетку вносятся две плазмиды, которые кодируют флуоресцентные белки. У них есть участки, которые будут деградировать, как и белки в клеточном цикле. Один из них экспрессируется в G1, потом деградирует на границе G1/S. Другой, наоборот, стабилизируется в S и деградирует в митозе, как циклин В.
Для прохождения G1-фазы нужны циклин D и CDK4 или циклин D и CDK6. Для вхождения в S-фазу — CDK2 и циклин E. Для координации экспрессии генов в G1/S нужен белок RB и семейство активаторов E2F. E2F — это транскрипционный фактор, который запускает транскрипцию белков, необходимых для S-фазы. Сначала он ингибирован за счет взаимодействия с супрессором RB, который его физически связывает и не дает ему активировать свои гены. RB монофосфорилируется комплексами CDK4/6 и циклина D. Это частично активирует E2F, и он активирует транскрипцию части генов, прежде всего циклина E. И вот уже циклин E в комплексе с CDK2 полифосфорилирует RB, в результате этот комплекс распадается, RB деградирует и E2F уже полноценно запускает синтез своих таргетных генов, прежде всего циклина A, который запустит репликацию. И затем активирующие E2F будут деградировать и заменяться ингибирующими E2F. Дальше идет репликация.
Семейство Myc
Еще один участник, который особенно важен для онкологии, — это c-Myc и все семейство транскрипционных факторов Myc. Это белки, которые регулируют множество генов, участвующих в клеточном цикле. Myc — один из самых ранних описанных онкогенов.
У него есть два партнера. Один называется Max. Вместе с Max Myc активирует транскрипцию почти всех генов, необходимых для продолжения клеточного цикла. Это и циклин D, и CDK 4, и E2F, и E3-лигазы. Комплекс Myc с другим партнером — Miz1 — репрессируют экспрессию ингибиторов клеточного цикла, за счет чего баланс смещается в сторону пролиферации.
Myc, как и циклин D, очень часто является таргетным геном, то есть большинство сигнальных каскадов увеличивает его экспрессию и стабилизирует его. Myc часто гиперстабилизирован в опухолях, иногда он амплифицирован, иногда просто его экспрессия увеличена, что связано с плохим прогнозом для пациента. Сейчас есть некоторые лекарства, которые могут снизить его активность, но они все находятся на очень ранних фазах разработки и пока не очень успешны.
Есть другой механизм, который особенно важен для вхождения в митоз, но также он активен на границе G1/S. Если клетка повреждена или испытывает стресс, то Wee1 будет фосфорилировать комплекс циклин-зависимой киназы 1 и циклина B, в результате его активность будет резко падать. Чтобы войти в митоз, нужна активность другой киназы, PLK1, которая ингибирует Wee1, активирует и стабилизирует CDC25. Крайне важной митотической киназой является PLK1, которая также необходима для сборки веретена деления.
Повреждение ДНК останавливает клеточный цикл
Повреждение ДНК часто активирует киназы CHK1 и CHK2. Они увеличивают активность Wee1 и фосфорилируют и деградируют CDC25, и клетка уже не продвинется дальше.
Из всех событий, которые активируют контрольные точки, самое важное — повреждение ДНК, оно активирует очень многие процессы. Целостность генома и опухолевая супрессия невероятно важны в многоклеточном организме.
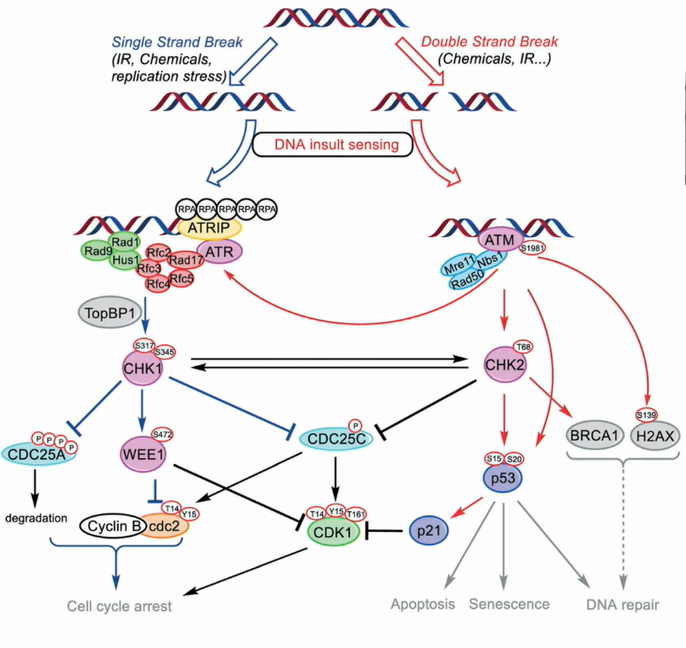
Двуцепочечные ДНК-повреждения активируют киназу ATM, а киназа ATM активирует киназу CHK2, которая фосфорилирует и стабилизирует белок p53, один из самых центральных опухолевых супрессоров. Это транскрипционный фактор, который может запустить клеточную гибель. Но если клетка не очень сильно повреждена, то он запускает транскрипцию p21, который блокирует циклин-зависимые киназы 1 и 2. А CHK2 будет деградировать CDC25 через ингибирующее фосфорилирование, и это смещает баланс в сторону ингибирующего.
Одноцепочечные повреждения, а также проблемы в репликации, например, торможение репликационных вилок, коллапс репликационных вилок или недостаток нуклеотидов, активируют киназу ATR, которая, в свою очередь, активирует киназу CHK1, фосфорилирующую и ингибирующую CDC25 и активирующую Wee1.
Клетка останавливается, если она повреждена, но она не может оставаться в таком положении вечно, она должна репарироваться, и тогда все эти сигналы уйдут. После этого клетка может продолжать деление. Если клетка не сможет репарировать повреждения, она должна или умереть, или выйти в сенецентное состояние, стабилизировать блок клеточного цикла и прекратить делиться.
Для онкологии очень важно состояние покоя, когда клетки не получают сигналов для начала деления или у них есть ингибирующий сигнал. Обычно это отсутствие факторов роста, плотные межклеточные контакты, потеря адгезии. В таких клетках увеличивается количество p21, p27, p57, снижается транскрипция, подавляется экспрессия других маркеров пролиферации, например, Ki67, и в принципе меняется транскрипционный профиль.
Это состояние играет ключевую роль в опухолевой супрессии. Клетки, особенно эпителиальные, при контакте друг с другом ингибируют деление, потому что у них увеличивается транскрипция и стабилизируется p27, и это называется контактным ингибированием. Чтобы опухоль вообще начала делиться, ей нужно это ингибирование преодолеть за счет мутаций в p27 или в промоторе p27, или должны произойти эпигенетические изменения. В опухолевой клетке p27 очень часто делетирован или ингибирован другим способом. С другой стороны, возможно, в клетке просто очень много циклинов и CDK, так что его просто не хватает. У организмов, устойчивых к опухолям, например, у голых землекопов, есть дополнительные механизмы, способствующие предотвращению деления за счет контактного ингибирования.
Клеточное старение
Клеточное старение — крайне интересный процесс, который происходит при повреждении клеток. Это может быть мутация, которая активирует онкогены, повреждение ДНК, просто раны или оксидативный стресс. Клетки не только прекращают делиться, они меняют свой фенотип, становятся больше, начинают выпускать цитокины, факторы роста и другие белки, которые будут помогать регенерации тканей вокруг них. Такие клетки очень долго сидят в ткани и поддерживают деление окружающих клеток.
У них есть много разных маркеров, обычно их красят на активность так называемой бета-галактозидазы, ассоциированной с клетчатым старением (senescence-associated β-galactosidase).
Несмотря на то, что этот процесс могут активировать совершенно разные состояния, в основном он завязан опять-таки на повреждение ДНК. У сенесцентных клеток увеличивается количество активных форм кислорода, которые повреждают ДНК еще сильнее. Таким образом, клетка становится все более и более сенесцентной. Чтобы не умереть, она увеличивает экспрессию антиапоптотических белков, таких как BCL-2, BCL-W, BCL-XL. Кроме того, клетка экспрессирует p21 и p16, которые блокируют деление, связывая комплексы CDK и циклинов.
Леонард Хейфлик показал, что клетка делится примерно 60 раз, и не больше. Потом оказалось, что это происходит из-за сокращения теломер, то есть участков на концах хромосом, которые могут быть реплицированы только с помощью специального фермента теломеразы. В основном в клетках теломеразы неактивны, поэтому теломеры сокращаются, через некоторое время это активирует ответ на ДНК-повреждение, и клетки становятся сенесцентными.
Из-за стрессов в организме накапливаются сенесцентные клетки, это считается довольно важным компонентом старения, однако не обязательным. Например, у лабораторных мышей стареющие клетки тоже накапливаются, но из-за частого деления. У них активные теломеразы во всех клетках и огромные теломеры: даже если выбить теломеразу, нужно шесть поколений, чтобы они начали преждевременно стареть. Тем не менее, такие лабораторные мыши тоже живут 2 года, то есть у них старение происходит не за счет укорачивания теломер.
Для опухолей это гораздо более важный вопрос, так как они быстро достигают предела деления. Поэтому в опухолях теломеразы, как правило, активны. Высокий уровень активности теломеразы является плохим прогностическим фактором для многих опухолей. Активацию ее экспрессии регулирует, например, c-Myc.
Еще один механизм, который активирует клеточное старение, — это мутации в онкогенах, которые могли бы превратить клетку в опухолевую. Обычно они увеличивают скорость деления настолько, что клетка, у которой еще есть работающие опухолевые супрессоры, просто повредит свою ДНК, а ответ на ДНК-повреждения сделает такую клетку сенесцентной.
Клеточное старение не только защищает нас от онкологических заболеваний, оно может и способствовать им, если сенесцентных клеток слишком много. В норме этот фенотип нужен прежде всего для ремоделирования тканей при повреждении, поэтому эти клетки выделяют факторы роста, которые подпитывает другие опухолевые клетки, тем самым способствуя онкогенезу и многим другим возрастным заболеваниям.
Если у старых мышей убить все сенесцентные клетки, то такие мыши не живут дольше, но зато они живут лучше. У них не наблюдаются многие признаки старения, например, катаракты. Поэтому исследователи начали искать вещества, которые бы убивали только стареющие клетки, —сенолитики.
Как клетка становится опухолевой
Представим себе изменения, происходящие в опухоли. Там должно быть больше белков, которые способствуют делению, и меньше белков, которые будут его подавлять. Циклины будут гиперэкспрессированы, у них будет увеличена стабильность. Прежде всего это касается циклина D1 и циклинов E и A. Особенно опасны амплификации циклина E, которые наблюдаются при раке молочного железа, яичника, легких. Ингибиторы, контролирующие контрольные точки, будут делетироваться, или их экспрессия будут уменьшаться эпигенетически. Часто теряется экспрессия белков CKI — потеря экспрессии p27 наблюдается в раках легких, молочной железы и мочевого пузыря, во многих опухолях человека изменен ген p16Ink4a путем делеции, точечной мутации или гиперметилирования. Происходит гиперэкспрессия CDC25 и подавление Wee1. Теряется экспрессия RB, p53 (50% случаев рака). Идет гиперэкспрессия или стабилизация c-Myc (в 60% опухолей).
Можно ожидать, что в любой опухоли, помимо мутаций в онкогенах, будут какие-либо мутации, затрагивающие контроль клеточного цикла. Ключевая черта опухоли — излишняя пролиферация. Первые химиотерапевтические лекарства просто били по всем делящимся клеткам. Это были повреждающие ДНК агенты. В 60-х годах были разработаны ингибиторы митоза, которые связываются с тубулином в веретене деления. Есть две основные группы — алкалоиды Винка (винкаалкалоиды, алкалоиды барвинка) и токсаны. Под их действием нормальное веретено деления не собирается. Эти лекарства, несмотря на высокую токсичность, очень эффективны. Они до сих пор используются во многих схемах лечения вместе с таргетной терапией или после таргетной терапии.
Возможные подходы к терапии
После того как были открыты механизмы регуляции клеточного цикла, люди начали пытаться разработать таргетные препараты, которые были бы направлены на конкретные белки, мутировавшие и гиперэкспрессированные в опухолях. Но со всеми таргетными препаратами есть проблема — опухоли часто могут их обходить, используя другие белки, которые выполняют ту же функцию.
В начале люди делали ингибиторы, которые били сразу по нескольким циклин-зависимым киназам. Но они оказались слишком токсичными, чаще всего потому, что влияли на циклин-зависимые киназы, нужные для транскрипции.
Все, о чем было рассказано выше, — это «немножко ложь или очень сильное упрощение», заметил докладчик. Естественно, что если бы клеточный цикл работал как часы, то ингибирование любой циклин-зависимой киназы останавливало бы клетки в той фазе, в которой они находятся. Но оказалось, что если нокаутировать циклин-зависимые киназы у мышей, то у нокаутных животных только CDK1 окажется необходимой для деления, все остальные необходимы только для некоторых тканей и взаимозаменяемы. В принципе, это понятно с эволюционной точки зрения, потому что дрожжи обходятся одним CDK1, она может заменить в большинстве клеток другие циклин-зависимые киназы. Поэтому опухоли могут обходить ингибирование циклин-зависимых киназ.
Есть еще два необычных подхода к терапии. Опухоли делятся быстро, и странным образом оказывается, что если они будут делиться еще быстрее, слишком быстро, то паровоз сходит с рельс. Есть ряд препаратов, которые направлены на то, чтобы убрать тормозящие механизмы, а не подавить деления. Такие препараты сейчас проходят клинические испытаниях, как раз против таких очень быстро делящихся опухолей, у которых репликационная машинерия на пределе.
Наконец, есть «подход синтетической летальности». Если есть два пути, которые друг друга заменяют, и один из них мутирован в нормальных клетках, можно заблокировать второй в опухолевых клетках; они погибнут, а нормальные клетки выживут.
Проблема покоящихся клеток
Не все опухолевые клетки пытаются делиться быстрее. Большой проблемой являются покоящиеся опухолевые клетки и опухолевые стволовые клетки. Опухоль гетерогенна, одни из ее клеток делятся быстро, другие медленно. Химиотерапия бьет по всем делящимся клеткам, а покоящиеся клетки хорошо ее переживают. У них есть и другие неприятные свойства. Например, они экспрессируют белки мультилекарственной устойчивости и откачивают во внешнюю среду противоопухолевые препараты.
Проблема в том, что мы убили 99% опухоли, то есть все быстрые делящиеся клетки, но покоящиеся клетки приспосабливаются к той химиотерапии, которую мы использовали. И если через десятки лет эта опухоль возвращается, то она уже будет устойчивой к терапии. Есть подходы, направленные на то, чтобы убить покоящиеся клетки, или выгнать их обратно в клеточный цикл.
Недавно группа Виктора Татарского опубликовала статью, в которой с помощью ингибиторов CDK8/19 ученые не давали клеткам уйти в блок клеточного цикла под действием иматиниба, препарата против хронического миелоидного лейкоза. Такие клетки быстрее погибали, потому что не могли остановиться в G1 фазе клеточного цикла и повреждались при репликации.
Гелина Копеина: «Без клеточной гибели терапия рака не сработает»

Вам будет интересно


 246
246
 0
0












