Меню
Меню





 Все темы
Все темы
Собери геном своего растения. О повторах, полиплоидии и трудностях аннотации
На конференции PlantGen2023, которая прошла в Казани 11–15 июля, Мария Логачева, старший преподаватель Центра молекулярной и клеточной биологии Сколтеха, сделала доклад «Сборка и аннотация геномов растений: проблемы и перспективы». Публикуем его в сокращенном виде. Какую роль в современной геномике растений играют нанопоровое секвенирование и Hi-C, зачем для анализа генома секвенировать РНК и, кстати, как продвигается сборка богатого транспозонами генома гречихи?

Среди наших публикаций к 50-й годовщине открытия двойной спирали было интервью с Марией Логачевой, в котором она рассказала, среди прочего, об упрямом геноме гречихи. Геном «завоеван» ретротранспозоном Athila — мобильным элементом, длина которого 10–15 тысяч нуклеотидов, то есть даже больше длины прочтения, которую дают инструменты Pacific Biosciences. Это крайне затрудняет качественную сборку. Теперь уже можно поговорить о том, каким образом удалось решить эту проблему (спойлер: с помощью платформы Oxford Nanopore). Однако многие растительные геномы все еще остаются «крепкими орешками».
Сложности геномики растений
Растения невероятно разнообразны, и столь же разнообразны их геномы. Они сильно варьируют по размерам — от 60 миллионов нуклеотидов, как у насекомоядных растений семейства пузырчатковых, до 150 млрд — это в 50 раз больше генома человека. Так же варьирует и количество генов — от менее 20 тысяч до более 100 тысяч у пшеницы. В сообществе биологов, в том числе и занимающихся биологией растений, существуют две контрастирующие точки зрения относительно сборки растительных геномов. Одни говорят, что это сложная задача, которую невозможно сделать без большого финансирования и большой команды биоинформатиков. Вторые ссылаются на то, что в последние годы произошло стремительное развитие методов секвенирования, и считают, что это уже простая техническая задача, которую можно перепоручать коммерческим компаниям — отправить ДНК и получить готовый результат. Я в своем докладе представлю консенсусную точку зрения, потому что в каждой из этих двух точек зрения есть некая правда.
С одной стороны, при современных технологиях теоретически должна быть возможна полностью правильная, совершенная сборка генома. Об этом говорит «Твиттерная теорема» — впервые ее написал в Твиттере, с трудом уместив в 140 символов, известный американский вычислительный биолог Юджин Майерс, автор программы BLAST и других, которыми пользуются биологи по всему миру. Он сказал, что полностью точная сборка генома должна быть возможна при соблюдении трех условий: ошибки секвенирования распределены случайно; распределение чтений подчиняется распределению Пуассона; чтения сильно превышают длину повторов. В этом случае даже высокая частота ошибок не мешает хорошей сборке. С другой стороны, в реальной жизни данные редко соответствуют этим условиям, поэтому, чтобы получить хорошую сборку, нужны определенные усилия как в «мокрой» части, лабораторной, так и в биоинформатических процедурах.
Факторы, которые мешают сборке генома, делятся на две группы: технические — проблемы с качеством данных, и биологические — свойства, присущие образцу, на которые экспериментатор никак повлиять не может. Проблемы с качеством данных — это, в первую очередь, ошибки секвенирования. Технологии, которые дают короткие чтения, довольно точные. Например, Illumina и MGI — у них ошибок мало, около 0,1–0,5%, но для сборки больших и сложных растительных геномов они практически не представляют ценности. Тут на передний план выходят технологии, которые дают длинные прочтения, — Oxford Nanopore Technologies и Pacific Biosciences. У них ошибок 3–5%, в сложных случаях 10%. И еще могут быть проблемы, связанные с контаминацией: в случае растений возможно присутствие симбионтов, эндофитных бактерий, — но до определенной степени можно их контролировать и снижать их влияние. Чего не скажешь о повторах.
Повторы — визитная карточка растений, они могут составлять до 95% генома. Существенная их часть обусловлена активностью мобильных элементов, в частности, ретротраспозонов. У растений существует несколько способов защиты от ретротранспозонов — подавление экспрессии, подавление трансляции. Но иногда все эти механизмы не срабатывают, и происходит «взрыв транспозонов», то есть бесконтрольное их распространение, приводящее к увеличению размера генома — и к большим проблемам со сборкой. Также у растений распространена полиплоидия, которая приводит к тому, что фактически весь геном становится одним большим повтором. И даже растения, о которых мы привыкли думать как о диплоидах, например, наш основной модельный объект арабидопсис, в ходе своей эволюционной истории претерпевали несколько циклов полногеномной дупликации — с последующим выкидыванием части генов, но многие гены остаются у них не однокопийными, а представленными большими мультигенными семействами. Это тоже осложняет сборку.
Второй фактор — гетерозиготность. Поскольку у большинства высших растений, за исключением мхов, основная часть жизненного цикла проходит в диплоидной стадии, секвенируем мы диплоидный геном, у которого есть отличия между отцовским и материнским гапломами. Гетерозиготность у эукариот очень сильно варьирует. У человека она невелика, а у некоторых объектов, у грибов, например, может доходить до десятков процентов; у растений гетерозиготность обычно в пределах 1–5%.
Как бороться с повторами
Наиболее существенный и наиболее понятный неблагоприятный фактор — повторы. С ними есть один сложный, но хорошо работающий рецепт: длина чтения должна быть больше, чем средняя длина самого распространенного типа повторов. Технологии Pacific Biosciences могут давать либо более короткие (10 тысяч), но более точные прочтения, либо более длинные, 40–50 тысяч, но менее точные — там ошибки доходят до 10 или даже 15%. Для Oxford Nanopore практически нет верхней границы. У энтузиастов-пользователей этой платформы есть неформальное соревнование, кто получит самое длинное прочтение, и в настоящее время рекорд где-то около 2,5 млн. Конечно, это требует особых подходов к выделению ДНК: необходима очистка от коротких фрагментов, иначе, если они присутствуют у вас в образце, на секвенирование в первую очередь пойдут именно они. Также критична чистота ДНК. Есть еще подход, основанный на коротких чтениях платформы Illumina, из которых делаются синтетические длинные чтения, но он совсем недавний и не был еще проверен на больших и сложных геномах.
Я приведу два примера сборки геномов, которые показывают, что на самом деле нет какого-то одного универсального решения. Хороший пример — геном борщевика Сосновского, Heracleum sosnowskyi, размер генома, оцененный по цитофлуориметрии, — 1,5–2 млрд нуклеотидов. Мы секвенировали его на Pacific Biosciences, получили около 3 млн чтений, примерно по 15 тысяч нуклеотидов. Получилась достаточно хорошая сборка, длина 1,6 млрд нуклеотидов — близко к полному размеру генома, N50 — 22 млн нуклеотидов. И показатель BUSCO хороший – это показатель полноты геномной сборки, оценивается наличие ультраконсервативных генов, какие должны быть у всех растений. Они делятся на С — complete, F— fragment, неполный, и М — missing, отсутствующий. Чем больше фракция С и меньше фракция М, тем лучше сборка. При этом С еще делится на S — single-copy, однокопийный и D — duplicated, дуплицированный. Доля D должна быть небольшая. То есть у борщевика с N50, и с полнотой сборки все хорошо.
А вот плохой случай — гречиха Fagopyrum esculentum. У нее размер генома похожий, мы получили похожее количество данных на Pacific Biosciences, и лучшее N50 сборки было 330 тысяч, на два порядка меньше. Мы не использовали эту сборку, потому что она была плохой по другим параметрам. В итоге пошла в публикацию сборка с N50 188 тысяч. Мы обратились к коллегам, которые занимаются мобильными элементами. Илья Киров нам помог, он посмотрел и сказал: «О, какие замечательные здесь ретротранспозоны!», мы сказали: «Какой ужас! Лучше бы их не было!». Это именно тот случай, когда произошел транспозоновый взрыв: у близкого к гречихе вида их сильно меньше. Поняв это, мы вернулись к задаче на основе другой платформы, на этот раз использовали Oxford Nanopore Technologies, максимизируя длину чтения. Нам удалось ее довести до 25 тысяч нуклеотидов, и тогда все хорошо собралось.
Гетерозиготность: два генома в одном
Фактор гетерозиготности тоже сильно осложняет сборку, потому что мы секвенируем смесь отцовского и материнского гапломов и хотели бы получить либо консенсус между ними, как обычно делается гаплоидный геном, либо полностью фазированный геном, то есть разобранный на материнскую и отцовскую хромосому. Но, как правило, мы получаем смешанную сборку, в которой часть регионов фазированная, а часть — сколлапсированная в гаплоидный. Такая сборка не просто бесполезна, а даже вредна, потому что ведет к ошибкам в биологических интерпретациях.
Как можно понять, что эта проблема присутствует в сборке? Здесь есть два ключевых показателя: можно предположить, что сборка смешанная, во-первых, если размер сборки больше, чем размер гаплоидного генома, который оценен по цитофлуориметрии, и во-вторых, если высока доля фракции D по BUSCO.
Как с этим можно бороться? До какой-то степени помогает поменять тип образца, использовать для секвенирования линию со сниженной гетерозиготностью, например, дигаплоид или самоопыляемую. Для гречихи мы так и сделали— гречиха вообще облигатно перекрестноопыляющаяся. Мы взяли самоопыляющуюся линию, и все стало гораздо лучше. И конечно, есть биоинформатические подходы — использование программ, которые находят такие участки у разобранных гапломов и коллапсируют их в консенсусную последовательность.
Вот это выглядело в нашей работе по борщевику: покрытие должно быть в районе 20, потому что это было наше среднее расчетное покрытие для этого генома. Для фракции S, однокопийных генов максимум покрытия в районе 20, а для фракции D, дуплицированных — в районе 10, то есть снижено в два раза. Это показывает, что большинство этих как бы дуплицированных генов — на самом деле просто несколлапсированный гаплом, поэтому на каждый из них приходится половинное покрытие. По метрикам BUSCO видно, что этих вариантов сборки очень много, высока доля фракции D. После применения специальной программы Mabs, разработанной Михаилом Щелкуновым, и доля D упала, и покрытие этой фракции нормализовалось, показывая, что теперь в сборке остаются не артефактные дупликации, а только реально дуплицированные гены.
Геном и хроматин
Но даже если у нас есть самые хорошие данные, сборка таких сложных геномов, как растительные, обычно не дает хромосомного уровня. Для этого нужны некоторые дополнительные данные, и нужна процедура, которая называется скафолдингом, — упорядочивание контигов, выстраивание их в более длинные последовательности — скафолды. В настоящее время стандартным подходом для скафолдинга является использование данных Hi-C — это метод для анализа конформации хроматина. Но это еще и замечательный способ улучшения и проверки геномных сборок, потому что самое большое число контактов происходит между участками, которые физически граничат друг с другом. И в случае геномной сборки мы можем решить обратную задачу: посчитать число контактов между разными контигами и, исходя из этого числа, вычислить расстояние между ними. В настоящее время это стандартный подход, он используется во всех масштабных геномных проектах, таких как DNAZoo, посвященный секвенированию геномов животных, у которых есть риск вымирания. И для растений то же самое применимо.
Hi-C можно и нужно использовать не только для построения сборки, но и для поиска ошибок в сборке. Например, участки в сборке стоят рядом, но между ними контактов по данным Hi-C нет, есть контакты с другими участками. Это указание на ошибку сборки; можно такой участок перенести, потом заново картировать и посмотреть, исправится ли ситуация.
Генетические карты
Еще один подход для построения скафолдов и для нахождения ошибок — использование информации из генетических карт. О нем я расскажу на примере сборки генома пастушьей сумки Capsella bursa-pastoris. Это растение семейства крестоцветных — недавний полиплоид, возникший в результате гибридизации видов Capsella orientalis и Capsella rubella. Оба этих вида обладают достаточно узким ареалом, тогда как сама Capsella bursa-pastoris — космополитное растение, одно из трех самых распространенных растений в мире. Поэтому оно является перспективным объектом для экологической генетики, для понимания того, как возникает высокая приспособленность.
Чтобы создать более надежную базу для таких исследований, мы собрали геном одной из линий Capsella bursa-pastoris из данных Pacific Biosciences, дополненных данными по генотипированию путем секвенирования на популяции F2, полученной от скрещивания этой линии, которую мы секвенировали, и некоей другой линии — контрастной, между которыми около 250 тысяч полиморфных позиций. Картировали эти данные о секвенировании F2 на контиги и посмотрели, как распределяются снипы. Мы видим у большинства растений либо гомозиготы, либо гетерозиготы, можем видеть рекомбинации в некоторых точках — это нормальная картина, когда все хорошо. А когда все плохо, распределение снипов выглядит по-другому: в случае ошибки сборки мы видим какую-то позицию в контиге, в которой очень много событий, выглядящих как рекомбинации в многочисленных индивидуальных растениях. Но понятно, что рекомбинация в одной и той же точке у очень многих растений независимо происходить не может, поэтому мы такие моменты трактовали как ошибку сборки — просто были объединены фрагменты генома, которые физически не граничат друг с другом, — и разрывали сборку по ним.
Помимо этого, генетические карты являются хорошим инструментом для скафолдинга, потому что они позволяют объединять контиги в группу сцепления и проводить скафолдинг с помощью того же Hi-C, только в пределах каждой группы сцепления, а не по всему геному. Это сильно уменьшает вычислительную сложность и повышает точность сборок.
Аннотация
После того, как сборка так или иначе сделана, предстоит еще одна задача — может быть, даже более сложная: аннотация. Она подразделяется на структурную, которая отвечает на вопрос «Что это такое, что здесь есть?» и представляет собой по факту разметку генома на отдельные элементы, их части, регуляторные элементы, повторы; и функциональную, которая отвечает на вопрос «Почему оно здесь и как оно работает?» Я в большей степени буду говорить про структурную аннотацию, потому что функциональная больше связана с экспериментами на конкретных генах.
Даже простой, казалось бы, вопрос: «Сколько генов у растений?» оказывается не таким простым. Это картинка из обзора под замечательным названием «Сколько генов есть у растений и зачем они там есть?» — обзор достаточно старый, но сейчас сохраняются те же закономерности. На ней показаны изменения представления о числе генов в двух самых популярных объектах — арабидопсисе и рисе Oryza sativa. У арабидопсиса оценка числа генов менялась на несколько тысяч, у риса еще сильнее — начиналось с более чем 50 тысяч, сейчас пришли к консенсусу, что около 40 тысяч. И если даже у таких хорошо изученных объектов аннотация несовершенна, то что же говорить обо всех остальных?
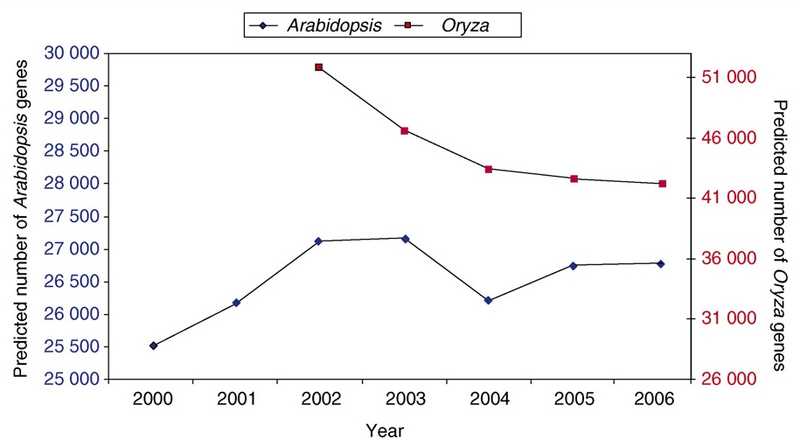 Current Opinion in Plant Biology 2007, 10:199–203. DOI: 10.1016/j.pbi.2007.01.004
Current Opinion in Plant Biology 2007, 10:199–203. DOI: 10.1016/j.pbi.2007.01.004
Следующий пример из работы Александры Касьяновой, посвященной пантранскриптому, — прежде чем приступать к этой работе, мы оценивали качество аннотаций для видов растений, которые в нее включены, и увидели, что для некоторых растений оно не очень хорошее. Параметр BUSCO для аннотированных генов не дотягивает даже до 90%. При этом сборка хорошая — полнота, оцененная по всему геному, везде больше 90%, а полнота, оцененная только по анонсированию генома, у некоторых видов меньше 90. И еще такой важный параметр, как процент одноэкзонных генов, — он достаточно консервативен у эукариот и должен составлять порядка 18–20%. У некоторых растений он очень сильно отклоняется от этой цифры, и это заставляет предположить, что многие гены были ошибочно аннотированы. И это очень популярные виды, сельскохозяйственно важные или модельные объекты. (…) Собирать геномы научились, а аннотировать пока еще нет.
Что мы можем с этим сделать, помимо того, что просто констатировать факт и огорчиться? Это зависит от задачи. В некоторых случаях какие-то большие сложные семейства генов необходимо просто аннотировать и размечать вручную. Есть несколько примеров для лейцин-богатых киназ или цитохромов, это огромные семейства генов с многими сотнями генов в них, здесь необходима ручная аннотация. Мне самой приходилось это делать, и всем рекомендую делать именно так в подобных случаях, а не полагаться на существующую аннотацию.
Если говорить о более глобальной разметке, не сфокусированной на отдельных генах, то здесь будут очень полезны данные секвенирования РНК. Можно напрямую секвенировать РНК на Oxford Nanopore, либо кДНК — ДНК, комплементарную РНК, как делали мы, и затем картировать на референсный геном. Например, картирование чтений кДНК на геном арабидопсиса показывает, что качество аннотации не очень хорошее даже для такого «золотого стандарта». Половина генов фактически не предсказана, чтение кДНК есть, а в геноме на этом месте ничего не размечено. Есть и менее глобальные, но тоже важные ошибки: проаннотирован интрон, и никаких изоформ не указано в аннотации, а по чтениям кДНК мы видим, что есть чтения с удержанием интрона. Так что длинные чтения, которые охватывают транскрипт целиком, полезны еще и для выявления новых изоформ.
Очень полезны специализированные транскриптомные данные, такие как CAGE, Ribo-seq, PAS-seq. Они позволяют найти старты и стопы транскрипции и трансляции, сайты полиаденилирования, и тоже пока не очень высокая точность в этом плане даже для арабидопсиса. CAGE — это секвенирование 5’-конца мРНК, и, например, можно видеть, что для гена есть три старта транскрипции, а в аннотации не размечен ни один из них.
Подводя итоги: сборка и аннотация геномов являются все еще сложной, но посильной задачей. Но для того, чтобы делать ее оптимальным образом, необходимо представлять возможности и ограничения технологий и обязательно использовать дополнительные данные в виде транскриптомов. Для этого оптимально подходит платформа Oxford Nanopore Technologies, так как сейчас у них появились высокопроизводительные приборы серии Promethion, которые дают десятки миллионов чтений, а очень высокая точность в данном случае не нужна.
Интервью подготовлено при поддержке компании SkyGen, дистрибьютора современных инструментов для секвенирования. Продукция, предлагаемая SkyGen, вызвала большой интерес на PlantGen2023.

Реклама. Рекламодатель ООО «Скайджин»
Токен: LdtCKFQDr
Вам будет интересно

 675
675
 0
0