Меню
Меню





 Все темы
Все темы
Генная терапия сегодня и в ближайшем будущем
Десятого октября 2024 года на платформе PCR.NEWS прошел вебинар «Генная терапия: новейшие технологии и перспективы развития». Читайте о российских подходах к генной терапии муковисцидоза, о том, как исправить мутации в митохондриальной ДНК, можно ли создать персональную терапию для маленького ребенка и какой терапевтический препарат сейчас самый дорогой в мире.
NIH | flickr.com | CC BY-NC 2.0
Участники вебинара рассказали о новейших отечественных и зарубежных разработках в сфере генной терапии заболеваний, связанных с мутациями в ядерной и митохондриальной ДНК, о разработке персонализированной генной терапии. Доклад представителя компании «Альгимед» был посвящен обеспечению приборной базы для проведения трансфекции.
Вебинар начался вступительным словом модератора Светланы Смирнихиной (ФГБНУ «МГНЦ им. академика Н.П. Бочкова»). Она отметила, что область генной терапии в России активно развивается. Это касается и технических аспектов, таких как методы доставки препаратов и редактирования генома, и экономических факторов — увеличивается доверие к области со стороны фарминдустрии, что способствует повышению финансирования. Растет и государственная грантовая поддержка исследований. Происходят изменения законодательства касательно персонализированных препаратов генной терапии, разработка которых ранее была невозможной. Модератор пригласила слушателей присоединиться к телеграм-каналу Gene therapy, в котором общаются многочисленные российские коллективы и публикуются анонсы мероприятий.
Светлана Анатольевна также представила первый доклад вебинара — «Коррекция патогенных вариантов в гене CFTR для лечения муковисцидоза». Работа по этой тематике ведется в Медико-генетическом научном центре имени академика Н.П. Бочкова и в данный момент выходит из стадии proof-of-concept на этап полноценных доклинических исследований. (Подробнее об этом Светлана Анатольевна рассказывала в своем интервью на PCR.NEWS.)
Муковисцидоз (МВ) — аутосомно-рецессивное заболевание, вызываемое мутациями в гене CFTR. Это одно из наиболее частых наследственных заболеваний в мире — 1 случай на 3000 человек, частота носительства, по разным данным, 1:23-1:40. Мутации в гене CFTR нарушают транспорт ионов хлора, что приводит к застою слизи в протоках респираторных путей, печени, поджелудочной железы и кишечника и провоцирует развитие воспалительных процессов. Продолжительность жизни с МВ составляет в среднем 37 лет.
Это заболевание относят к моногенным, однако спектр мутаций, приводящих к МВ, достаточно широк. Выделяют шесть классов мутаций по тому, на какие функциональные составляющие ионного канала они воздействуют.
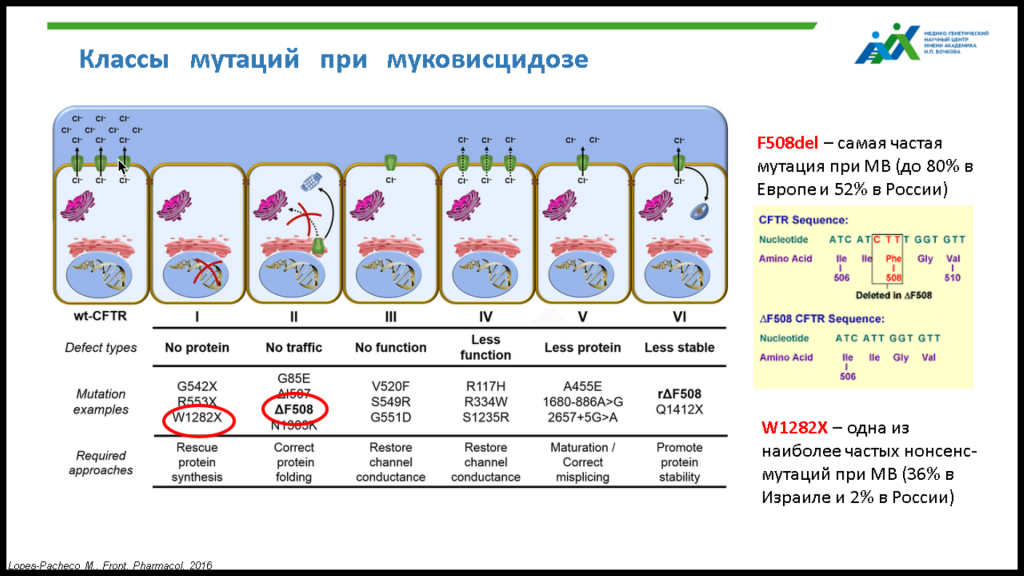
Так, при мутации W1282X, относящейся к наиболее тяжелому первому классу, отсутствует сам белок ионного канала. При мутациях второго класса, например, ∆F508, белок образуется, но имеет неправильную структуру и быстро деградирует. Начиная с третьего класса мутаций белок выходит на апикальную поверхность клетки, но не функционирует либо функционирует плохо. Коллектив Светланы Анатольевны сосредоточился именно на мутациях W1282X и ∆F508 как наиболее распространенных (2% и 52% мутаций у больных в РФ) и тяжелых.
В настоящее время методы традиционной терапии МВ представлены потенциаторами, повышающими функцию белка на поверхности клетки, и корректорами, способствующими транспорту белка к поверхности и исправляющими дефекты в его процессинге. Пациенты с нарушением синтеза белка пока что остаются без лечения. С 1993 года разные группы ведут клинические исследования по генозаместительной терапии МВ, но до сих пор ни одно из них не показало достаточной эффективности, чтобы получить одобрение на клиническое использование.
С 2016 года группа Светланы Анатольевны разрабатывает генную терапию МВ путем коррекции мутаций с использованием CRISPR-Cas систем, об эволюции которых спикер коротко рассказала.
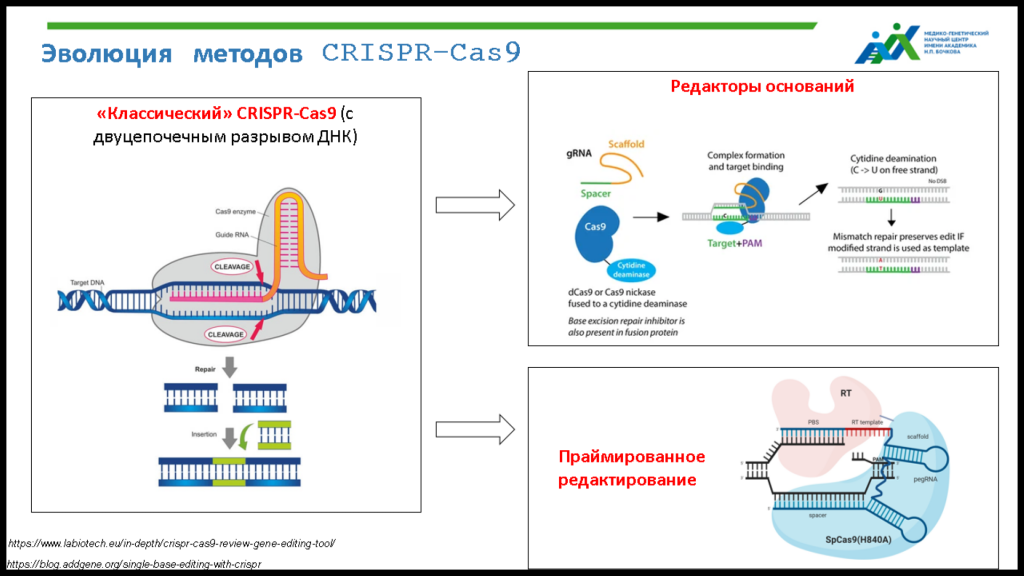
Начинала группа свою работу с «классического» CRISPR-Cas9, затем перешла на более поздние модификации (редакторы оснований и праймированное редактирование), которые не создают двуцепочечных разрывов ДНК и используют вспомогательные белки для замены оснований, что снижает риск офтаргетных изменений в геноме и создания новых мутаций в процессе редактирования. «Классический» CRISPR-Cas9 и метод праймированного редактирования применялись для исправления мутации ∆F508, а редакторы оснований (способные исправлять только точковые мутации) — для коррекции мутации W1282X.
Все работы в лаборатории ведутся на клеточных культурах, полученных из фибробластов кожи пациентов с МВ, которые наблюдаются в НИИ пульмонологии ФМБА России у Елены Львовны Амелиной. С помощью направленного репрограммирования из фибробластов получают индуцированные плюрипотентные стволовые клетки (иПСК), которые затем дифференцируют в легочные и бронхиальные органоиды и базальные клетки легкого. Преимущество последних — способность к саморегенерации и дифференцировки в множество типов клеток легкого. Редактирование мутаций в базальных клетках, отвечающих за восстановление эпителия, теоретически позволяет получить долгосрочный эффект после однократного введения препарата. По этой причине, а также из-за большего удобства работы с 2D структурами, все опыты проводились именно на культурах базальных и индуцированных плюрипотентных стволовых клеток.
Первичные опыты с «классическим» CRISPR-Cas9 позволили достичь определенной эффективности редактирования мутации ∆F508, которая, однако, сильно варьировалась (от 3% до 30% в зависимости от пациента, типа клеток и используемой комбинации эндонуклеазы и ДНК-«заплатки»). При этом по разным опубликованным данным для восстановления нормального состояния легких достаточно отредактировать от 6–10% до 20% клеток. Данную отметку классический метод преодолевал лишь в отдельных случаях.
Для применения праймированного редактирования был проведен скрининг 24 вариантов редактирования на эффективность исправления мутации ∆F508 у двух пациентов. Шесть вариантов преодолели порог в 10% отредактированных клеток, но только для одного из двух больных. По словам докладчицы, высокая вариабельность между пациентами наблюдается в исследованиях до сих пор, и группа работает над устранением этих различий.
В лаборатории также был отработан метод создания из базальных клеток 3D органоидов легкого, исследование которых позволяет подтвердить, что редактирование мутантного аллеля восстановило функции ионного канала. Для этого к культуре добавляют форсколин, который при нормальной работе CFTR канала вызывает набухание органоида. Для ряда вариантов редактирования наблюдалось значимое увеличение объема органоидов в сравнении с контрольными мутантными клетками, то есть ионный канал работал нормально.
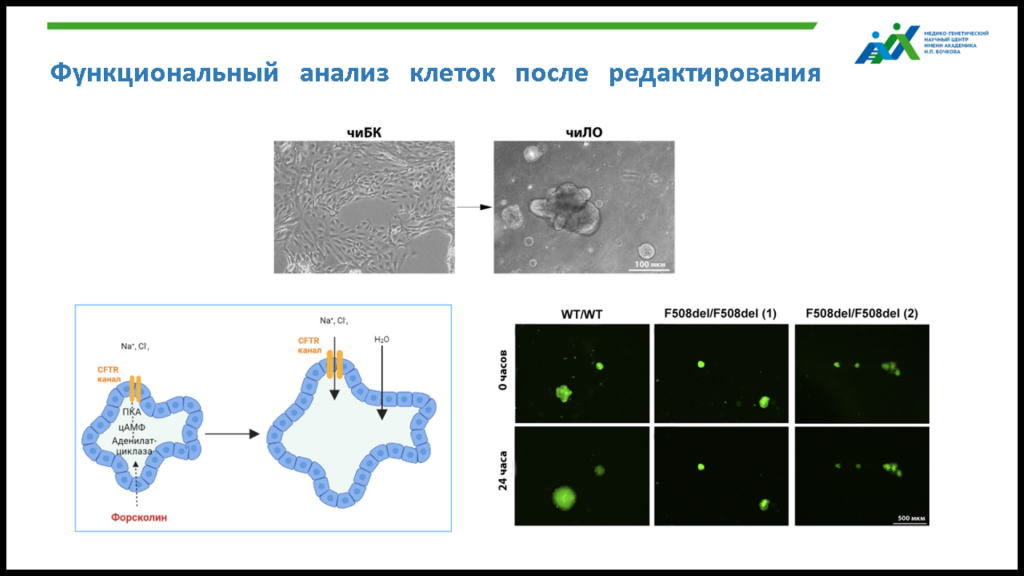
Редактирование точковой мутации W1282X проводилось при помощи аденинового редактора оснований, конвертирующего таргетный аденин в гуанин. Данный метод показал эффективность редактирования около 10% на иПСК, однако эффективность на базальных клетках оказалась ниже (около 6%). На базальных клетках также наблюдалось редактирование нецелевого аденина, находящегося в соседней позиции с таргетным локусом, с частотой около 1% (достоверно выше, чем в контрольной группе).
Таким образом, на данный момент наибольшую эффективность (около 30%) показало редактирование мутации ∆F508 методом праймированного редактирования базальных клеток легкого. Это направление будет разрабатываться в рамках доклинических исследований. Методы редактирования мутации W1282X перед переходом к следующим этапам исследования требуют доработки для повышения эффективности.
Отвечая на вопросы, Светлана Анатольевна рассказала, что для проведения подобных исследований в лаборатории необходимо иметь оборудование для работы с прокариотами, эукариотическими клетками человека, проточной цитометрии, крайне желательно наличие клеточного сортера. Также необходим генетический анализатор (эта часть работы может проводиться на аутсорсе) и возможности для глубокого таргетного секвенирования нового поколения. Докладчица отметила, что если при воздействии на базальные клетки теоретически можно обойтись однократным приемом препарата, то при таргетировании эпителиальных клеток легкого будут необходимы повторные приемы препарата каждые полгода-год. На вопрос о том, какое ПО используется для анализов данных NGS, Светлана Анатольевна ответила, что основной инструмент — бесплатная программа CRISPResso2.
Второй доклад вебинара, «Клинические перспективы исправления мутаций ДНК митохондрий человека», представил Илья Мазунин (Центр молекулярной и клеточной биологии Сколковского института науки и технологий). Доклад имел обзорный характер и был посвящен инструментам и подходам к генной терапии митохондриальных заболеваний в контексте их клинического применения в ближайшей перспективе.
Илья Олегович сравнил редактирование генома митохондрий с операцией на сердце плода в утробе матери, которая требует особых подходов в сравнении с простой операцией на сердце. Так и работа с геномом митохондрий значительно отличается от редактирования ядерного генома.
Митохондриальная ДНК кодирует 22 транспортных РНК, две рибосомных и 11 матричных РНК, которые транслируются в 13 субъединиц комплекса окислительного фосфорилирования. В то же время множество генов, которые кодируют белки, участвующие в метаболизме митохондрий, находятся в ядре и являются предметом «классического» генного редактирования.
Механизмы возникновения мутаций в митохондриальной ДНК не отличаются от таковых в ядерной ДНК: прежде всего это ошибки репликации (около 58% мутаций) и повреждения ДНК (42%). Около 20% мутаций представлены специфическими заменами, связанными с дезаминированием аденина.
Еще одним частым видом мутаций мтДНК являются делеции. Группа Ильи Олеговича предложила модель, согласно которой сама нуклеотидная последовательность мтДНК способствует выпадению фрагментов в результате взаимодействия двух идентичных локусов (прямых повторов). С делециями связано множество митохондриальных заболеваний, таких как синдром Пирсона и наружная офтальмоплегия. Докладчик обратил внимание слушателей на то, что в клетке находятся до нескольких тысяч молекул мтДНК, и невозможно возникновение мутаций во всех одновременно — мутации возникают в единичных молекулах и далее распространяются. Поэтому клинические проявления могут возникать не сразу, а лишь через несколько поколений потомков.
Илья Олегович привел список основных систем редактирования митохондриальной ДНК, включая нуклеазы (mitoARCUS, mitoRE, mitoTALEN, mitoZFN, и mitoCas9 с mito-sgRNA) и редакторы оснований (DbCBE, mitoZFD и sTALED).
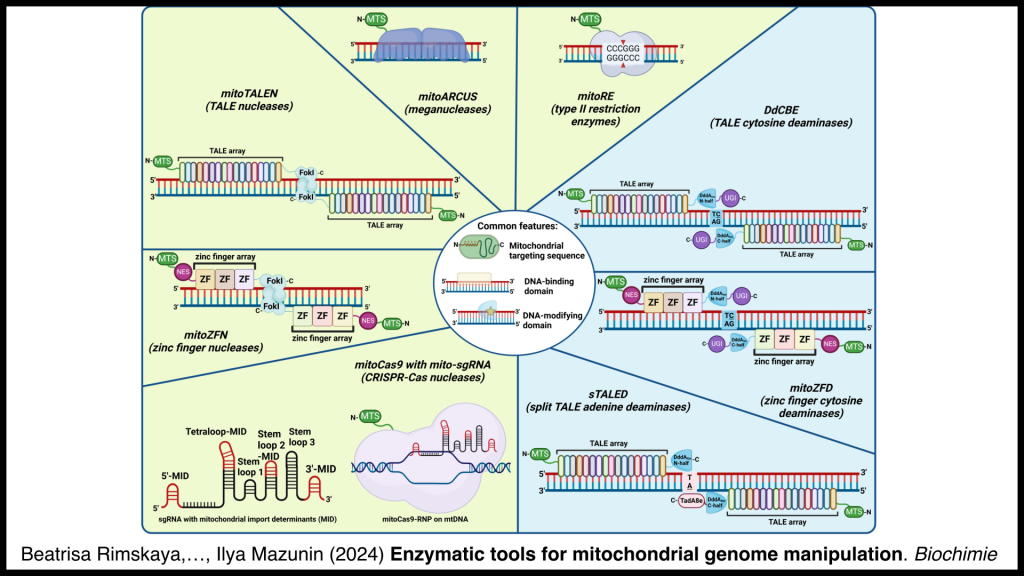
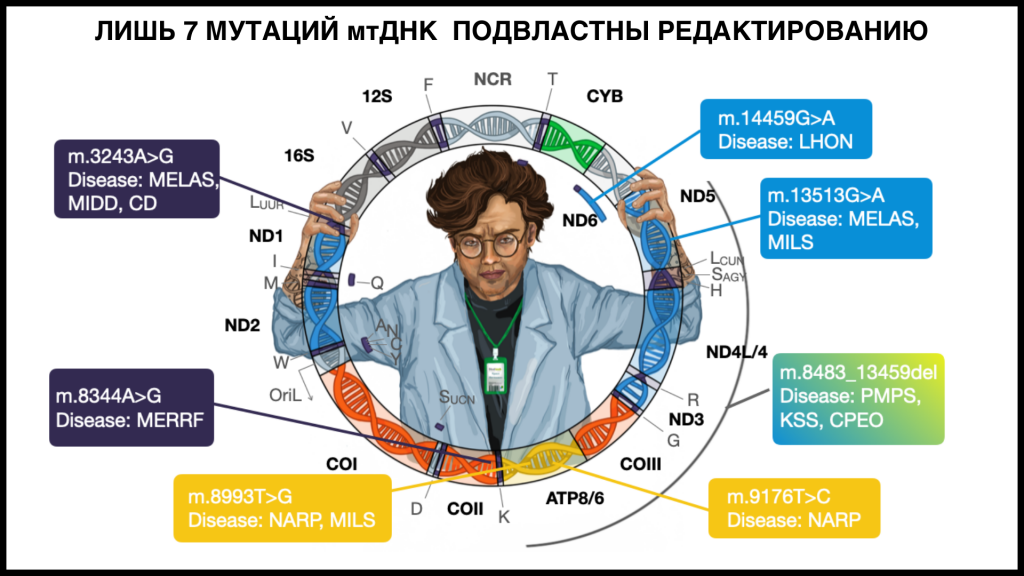
Несмотря на широкий арсенал методов редактирования, из 96 подтвержденных патогенных мутаций мтДНК до сих пор лишь семь удалось отредактировать на практике; в теории возможно редактирование 86 мутаций. Все семь были исправлены путем внесения двуцепочечных разрывов в мутантную ДНК и ее дальнейшей деградации. В отличие от ядерной ДНК, в митохондриях невозможно починить двуцепочечный разрыв с помощью гомологичной рекомбинации или негомологичного соединения концов, мутантная мтДНК с разрывами просто деградируется специальными ферментами.
Особое внимание докладчик уделил системе ARCUS — запатентованной технологии компании Precision Biosciences. Система основана на перекодировании мегануклеазы под специфический таргетный локус. (PCR.NEWS писал о ней в 2022 году.) В доклинических работах, проведенных под руководством одного из пионеров генной терапии, доктора Карлоса Мораеса из университета Майами, ARCUS показал высокую специфичность в гидролизе мтДНК с мутацией m.3243G, ассоциированной с целым спектром патологий, включая нейродегенеративный синдром MELAS. В рамках данных исследований не наблюдалось офтаргетных взаимодействий белка с ядерной ДНК. Система также показала работоспособность in vivo на моделях ксенографных мышей (линии мышей с такой мутацией не существует, поэтому редактировали ксенотрансплантат, содержащий мутантные клетки). Эффективность была ниже, чем на культурах клеток, но причиной могла быть инкапсуляция ксенографа, затрудняющая доставку препарата.
Терапия на основе редактирования или элиминации мутантных мтДНК пока не прошла дальше этапа исследований на животных моделях, однако испытания заместительной терапии с использованием аденоассоциированных вирусных векторов (AAV) уже проводятся на людях.
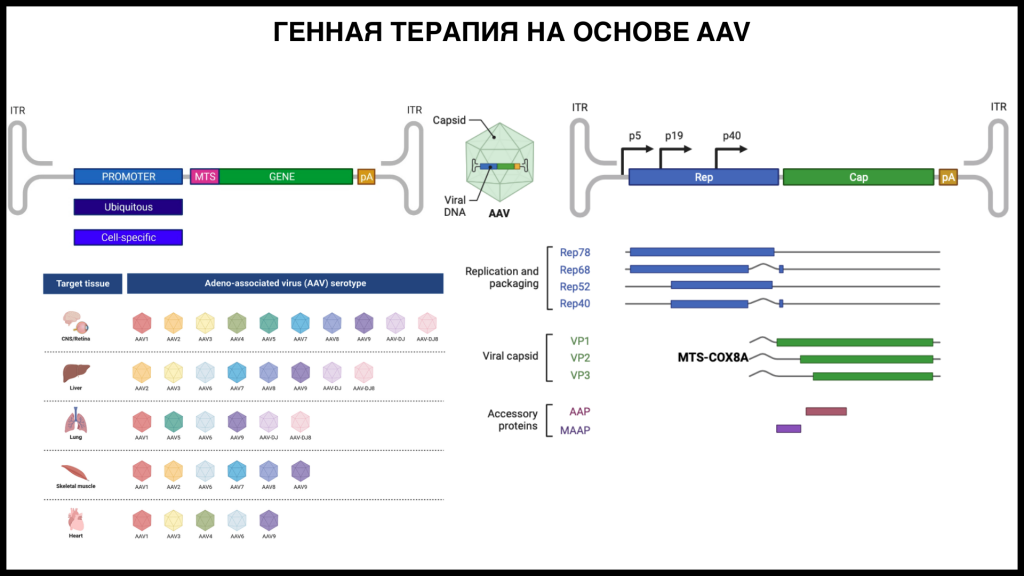
Заместительная терапия заключается в доставке в митохондрии «правильных» вариантов мутантных генов. При этом существуют два подхода. В первом случае AAV доставляет в клетку митохондриальный ген без мутации, кодоноптимизированный под трансляцию на рибосомах цитоплазмы; белок транслируется в цитоплазме и затем транспортируется в митохондрии. Во втором случае один из капсидных белков AAV модифицируется — к нему добавляют сигнал митохондриальной локализации; вирусные частицы проникают внутрь митохондрий, где и происходит трансляция белка. Добиться успешного применения второго подхода у группы Ильи Олеговича не получилось, хотя этот подход показал эффективность в ряде клинических исследований.
В качестве примера успешного применения обоих подходов докладчик привел опубликованные результаты клинических испытаний генной терапии оптической нейропатии Лебера, проведенных двумя группами ученых — в Китае и США. Всего в настоящее время завершено два клинических исследования этого заболевания, три находятся в активной фазе и одно остановлено в связи с потерей финансирования. Одобренных к применению препаратов пока не имеется.
В заключение Илья Олегович отметил, что он ожидает начала применения препаратов генной терапии в обозримом будущем.
Следующий доклад под названием «Виротерапия наследственных и онкологических заболеваний» представил Александр Карабельский из Научного центра трансляционной медицины университета «Сириус».
Докладчик кратко рассказал о центре трансляционной медицины, в котором существуют два направления — генная терапия, за которую отвечает Александр Владимирович, и медицинская биотехнология, которой руководит Роман Алексеевич Иванов. Отделение генной терапии занимается преимущественно вирусными векторами, в то время как направление медицинской биотехнологии включает в себя разработки на основе рекомбинантных белков и малых молекул. Таким образом, университет охватывает почти все основные технологические платформы генной терапии.
Александр Владимирович напомнил основные вехи истории генной терапии — от фундаментальных генетических открытий в начале XX века к первым пациентам, получившим генную терапию в 90-х годах XX века, и до современных разработок.
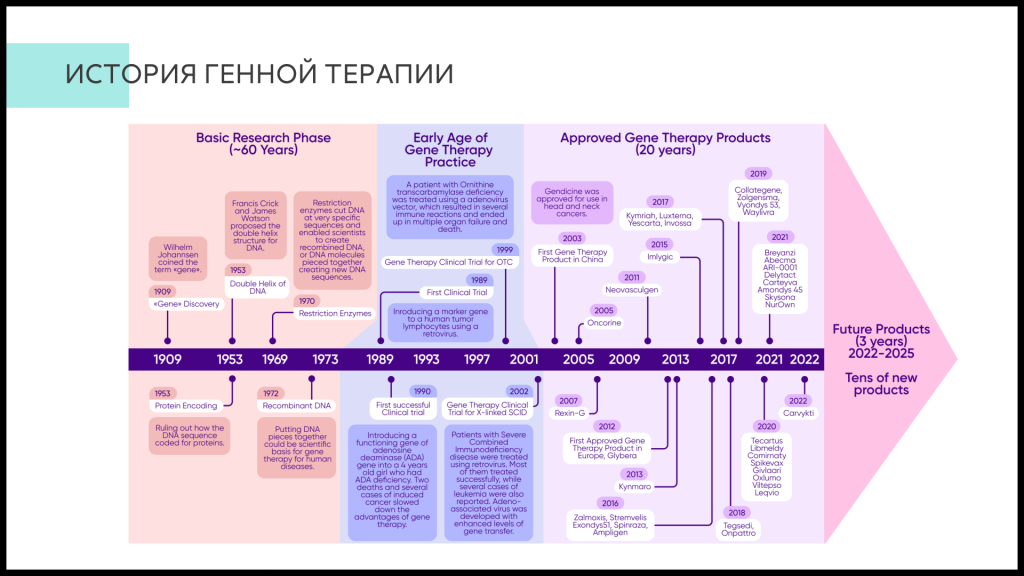
В настоящее время ежегодно регистрируется 3–5 продуктов по генной терапии. Так, за последние три года вышел целый ряд продуктов на основе AAV для терапии дефицита декарбоксилазы ароматических аминокислот (65–70%-ное восстановление двигательных функций), гемофилии А (97,5% не нуждались в введении FVIII) и гемофилии В (94% не нуждались в введении FIX), синдрома Дюшенна. Последний, правда, требовал введения большой дозы препарата, связанной с сильными проявлениями токсичности. Отдельно докладчик остановился на комбинированной терапии бета-талассемии и серповидноклеточной анемии, в которой применяется ex vivo модификация стволовых клеток пациента, отключающая ингибирование экспрессии фетального гемоглобина (подробнее о результатах фазы 1-2 клинических исследований на PCR.NEWS).
В рамках направления «Генная терапия» университета «Сириус» разрабатываются проекты по генозаместительной терапии различных форм наследственной слепоты, созданию онколитических вирусов; в 2024 году запущена разработка технологий невирусной доставки нуклеиновых кислот.
Онколитические вирусы — относительно новый класс препаратов; это вирусы, способные реплицироваться в раковых клетках, но при попадании в здоровые клетки они элиминируются механизмами противовирусного ответа. Онколитические вирусы могут не только вызывать лизис раковых клеток, но и синтезировать в них определенные белки, направленные на активацию противоопухолевого иммунного ответа, который будет подавлять и рост метастазов вдали от опухоли.
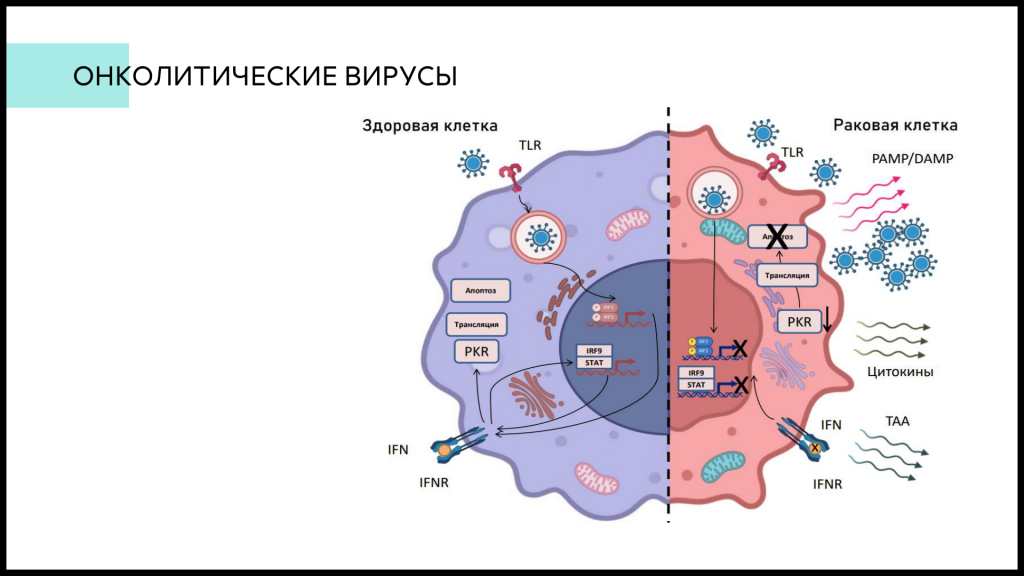
Группа Александра Владимировича занимается разработкой подобных препаратов на основе вируса везикулярного стоматита, в который встраиваются гены активации иммунитета. Докладчик описал трудоемкий процесс «оживления» вируса — клеточная линия трансфицируется смесью плазмид, что приводит к сборке вирусной частицы, которая далее реплицируется и очищается. Первый этап наиболее критичен, так как далеко не всегда получается добиться успешной сборки вирусной частицы.
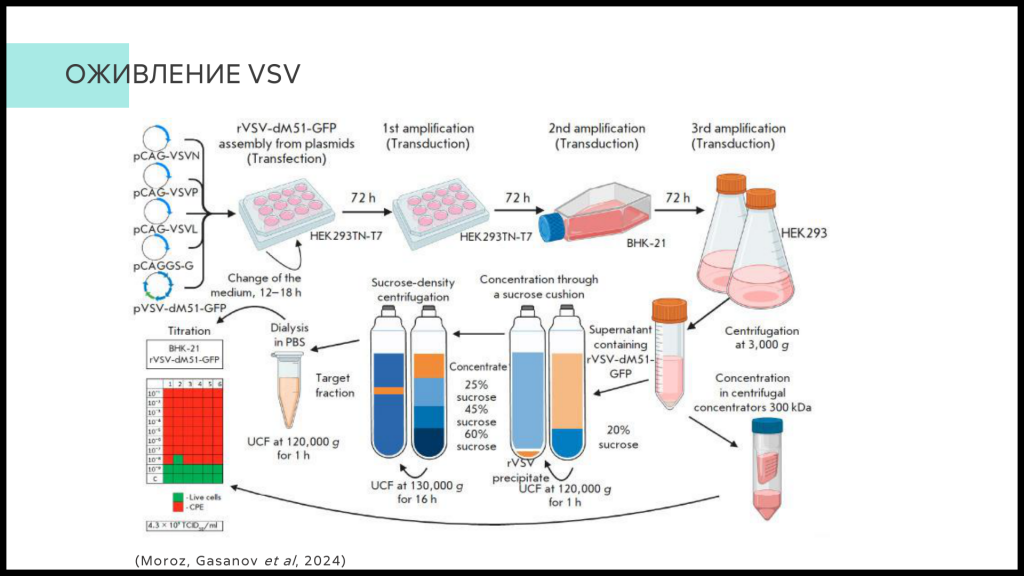
В рамках нового гранта группа работает над комбинацией вирусных частиц с липосомными частицами, несущими генетический материал, кодирующий иммуностимулятор. Такой вынос модификаций в отдельную частицу может позволить более надежно синтезировать препарат, пропуская нестабильный этап синтеза модифицированного вируса.
Активно идет процесс подбора моделей для in vivo тестирования препаратов, так как для проверки действия иммуностимуляторов требуются лабораторные животные с рабочим иммунитетом, а не иммунодефицитные, как обычно в экспериментальной онкологии. На моделях меланомы и легочной карциномы была показана биологическая активность вируса. Также удалось показать, что эффективность репликации вируса существенно различается в разных клеточных культурах.
Для терапии глазных болезней в университете разрабатываются подходы на основе классической генозаместительной терапии с использованием рекомбинантных AAV векторов, которые вводят в стекловидное тело либо под сетчатку глаза. Александр Владимирович отметил, что все AAV-препараты в настоящее время подвергаются тем или иным модификациям — например, выполняется оптимизация кодонов терапевтического гена или добавление регуляторных элементов.
Группа Александра Владимировича работает с вирусами AAV5, AAV6, AAV8, AAV9 и AAVDJ, на основе которых разрабатываются препараты, таргетирующие гены RDH12 (мутации в нем вызывают амавроз Лебера 13 и пигментный ретинит), PDE6B (дистрофия сетчатки и пигментный ретинит) и AIPL1 (амавроз Лебера 4).
Продукт гена AIPL1 — кошаперон, участвующий в фолдинге белков с нестандартными посттрансляционными модификациями, в том числе PDE6B. Так как AIPL1 — достаточно нестандартный белок, группа решила изучить эффекты от его экспрессии в клетках. Для этого была получена кодоноптимизированная версия гена, которая затем экспрессировалась в клетках линии ARPE-19. Оказалось, что экспрессия гена дикого типа активировала иммунный ответ по ряду генов, в то время как для кодоноптимизированной версии иммунной активации не наблюдалось. Это очень важные результаты, так как активация противовирусного ответа может снижать эффективность терапии.
Проводятся также модификации серотипа вируса, позволяющие повысить эффективность трансдукции в сотни раз в сравнении с вирусами дикого типа. Это может быть, например, модификация поверхностной петли оболочки вируса специальным пептидом.
Наконец, возможна модификация последовательности белка, обеспечивающая его транспорт в митохондрии. Такие модификации необходимы, например, при терапии оптических нейропатий, связанных с мутациями в гене ND4. Группа Александра Владимировича занимается разработкой препарата с аллотопической экспрессией гена — белок, модифицированный сигналами митохондриальной локализации, экспрессируется в цитоплазме и далее транспортируется в митохондрии. У исследователей уже получилось снизить в клетках уровень активных форм кислорода (которые накапливаются при мутациях в ND4).
Докладчик также остановился на проблеме создания животных моделей ретинопатий, вызванных мутациями в митохондриальном геноме. Проблема связана со сложностью индуцирования единичных мутаций, а также с наличием компенсаторных механизмов у животных. Один из возможных подходов включает в себя инъекцию вектора, несущего мутантный вариант гена, чтобы «испортить» митохондриальный генотип, а затем введение терапевтического вектора для восстановления функции.
В завершение Александр Владимирович перечислил темы, входящие в новый грант по невирусной доставке нуклеиновых кислот. Они включают в том числе терапию гемофилии Б и ретинопатии, праймированное редактирование, комбинированные терапии онкологических заболеваний.
После доклада обсуждались вопросы генно-клеточной терапии с трансплантацией пигментного эпителия, которой посвящены пока единичные исследования. По словам Александра Владимировича, такой подход потенциально весьма эффективен, но требует высокой степени персонализации.
Четвертый доклад, «Персонализированная генотерапия», сделал директор института трансляционной медицины НМИЦ АГП им. В.И. Кулакова Денис Ребриков.
В России около 1% новорожденных (15000 детей в год) рождаются с моногенными наследственными заболеваниями. Из тех, кто страдает от редких заболеваний, 30% не доживает до 5 лет. Каждый день в России от моногенных наследственных заболеваний умирают 10 детей.
В Центре Кулакова уже три года функционирует проект скрининга новорожденных методом полного экзомного секвенирования ЭКЗАМЕН — Экзомный Клинически Значимый Анализ Мутаций Единичных Нуклеотидов (о некоторых его предварительных итогах на PCR.NEWS). В рамках проекта было собрано 13000 образцов пуповинной крови. Для 11000 из них был получен полный экзом. По данным первых 7000 новорожденных была опубликована статья: 3% новорожденных имели серьезные генетические заболевания. Такую высокую долю докладчик объяснил тем, что в центре наблюдаются преимущественно беременные женщины с высоким риском, в том числе и генетических нарушений.
Скрининг на патогенные варианты проводился по 2350 генам. У 0,9% фенотипически нормальных младенцев были выявлены клинически значимые варианты, связанные с заболеваниями с ранним началом, поддающимися своевременному лечению (среди детей с явными признаками болезни это значение превысило 10%). У 2,1% были выявлены варианты, ассоциированные с пониженной пенетрантностью, моногенными заболеваниями с поздним началом, заболеваниями с переменной экспрессивностью. У 0,3% — хромосомные аномалии.
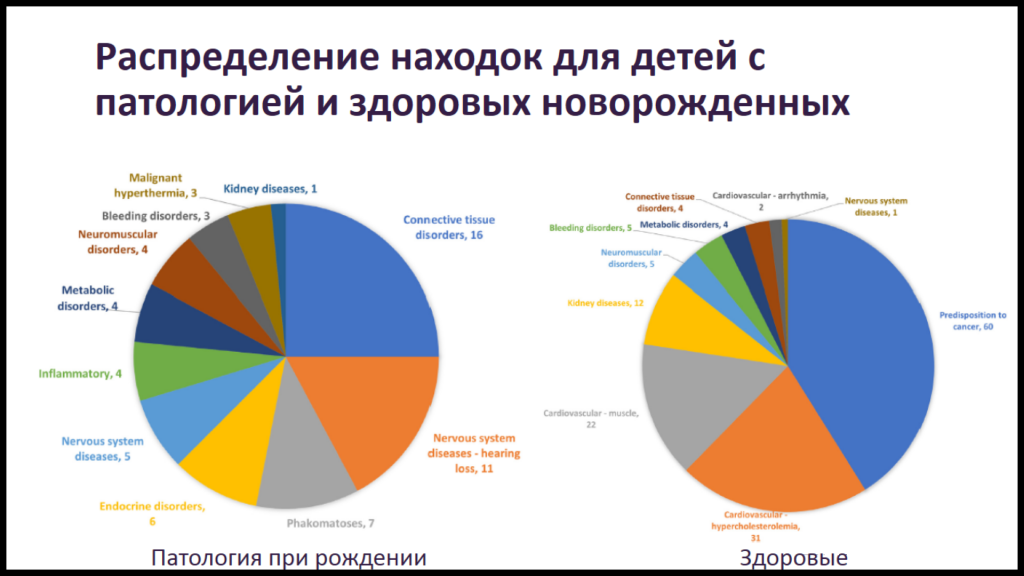
Таким образом, даже в одном медицинском центре регистрируются сотни различных генетических нарушений. В то же время в мире одобрено менее 20 генотерапевтических препаратов, еще около 30 находятся на фазе 3 клинических исследований. Это связано в том числе с нерентабельностью инвестиций в разработку препаратов для терапии редких заболеваний.
По мнению Дениса Владимировича, около 6000 заболеваний могут никогда не получить зарегистрированный генотерапевтический препарат. Это около половины детей, рождающихся с моногенными заболеваниями, или около 8000 детей в год на территории РФ. Отсюда острая необходимость в разработке препаратов под конкретного пациента. Проведение скрининга новорожденных позволяет начать разработку терапии еще до проявления симптомов. Однако современный подход к регистрации препаратов не подходит для подобных разработок, поэтому требуется модификация законодательства. Контролировать необходимо процессы изготовления и применения препарата, а не сам препарат, отметил докладчик.
Цена одной дозы индивидуального препарата без регистрации составляет около миллиона рублей, а срок изготовления — от одного до трех месяцев (три месяца нужны, если проводятся испытания на животных). Этот срок включает подбор праймеров и наработку генов (семь дней), сборку конструкции (семь дней), упаковку вируса и его очистку (14 дней) и доклинические испытания (30–60 дней). При этом клинические признаки болезней с ранним началом обычно проявляются в возрасте от полугода до двух лет.
Для реализации персонализированной терапии необходим механизм доступа к применению генотерапевтического препарата по принципу Hospital Exemption, разрешающий применение препаратов, разработанных на базе медицинского учреждения для персонализированной терапии, без стандартного процесса регистрации. Такой подход уже принят в странах Евросоюза.
В настоящее время разработка персонализированных препаратов включает следующие этапы:
-
получение заявки от родителя;
-
проверка по критериям;
-
изготовление препарата;
-
доклинические испытания на безопасность;
-
введение препарата.
Для включения в программу случай должен удовлетворять следующим критериям:
-
моногенное рецессивное заболевание;
-
не летальное в ближайшие два года либо обладающее альтернативным методом лечения;
-
открытая рамка считывания менее 4 т.п.н.;
-
наличие согласия родителей;
-
одобрение этическим комитетом и врачебной комиссией.
Документарное сопровождение включает протокол клинического исследования, информационный листок участника клинического исследования, форму информированного согласия, сообщение спонсора в этический комитет, заключение комитета и страхование клинического исследования.
Денис Владимирович привел пример функционирования подобного алгоритма на синдроме Криглера–Найяра — заболевания, связанного с нарушением функции гена UGT1A1, который кодирует белок, участвующий в конъюгации билирубина в печени. У больного накапливается билирубин в крови, что проявляется в виде желтухи и ведет к тяжелым поражениям мозга. Заболевание поражает примерно одного младенца из миллиона. Традиционные методы терапии — фототерапия, плазмаферез и трансплантация печени.
Была создана стандартная генетическая конструкция на основе AAV вектора для направленной экспрессии гена в печени. Безопасность проверили на мышиных моделях. Препарат был введен пациентке семи лет, которая до этого получала 12 часов фототерапии в день. После первого введения препарата уровень билирубина в крови уменьшился примерно в два раза, что позволило снизить время фототерапии до 4 часов в день. Однако в последующие полгода уровень билирубина плавно возрастал. После повторного укола (это был первым случаем повторного введения одного и того же препарата) также наблюдалось падение уровня билирубина, но после полной отмены фототерапии он резко вырос. Фототерапию возобновили в режиме «два часа в день», что позволило стабилизировать уровень билирубина на более низких значениях, чем до начала генотерапевтического лечения, и он остается таким низким около года со второго введения препарата.
Следующая часть доклада была посвящена этическим проблемам скрининга новорожденных и роли персонализированной терапии в их решении. Например, ребенок фенотипически здоров, но через месяц по результатам экзомного исследования у него обнаруживается тяжелое наследственное заболевание с началом в два-три года и высокой смертностью около пяти лет (данные цифры взяты для примера, подчеркнул докладчик). Могут ли исследователи сообщить родителям об этом? Если нет персонализированной терапии, то оба решения — сообщить или не сообщить — этически небезупречны. Возможность проведения персонализированной терапии позволяет сразу предоставить родителям вариант лечения (хоть и без гарантий), которое можно начать еще до проявления симптомов.
С целью внедрения персонализированных терапий Минздрав начал строительство двух комплексов — для GMP-производства биомедицинских клеточных продуктов на территории НМИЦ АГП имени Кулакова и для GMP-производства генотерапевтических препаратов — на территории РНИМУ. Реализацию проектов планируется завершить к 2026 году, с началом полноценного выпуска (200 препаратов в год) в 2027 году.
В заключительной части доклада Денис Владимирович обсудил критерии неонатального скрининга в контексте развития персонализированных терапий. В настоящее время в России придерживаются международных правил:
-
наличие четких клинических и биохимических критериев у заболевания;
-
известная частота заболевания;
-
заболевание без адекватного или своевременного лечения приводит к серьезной потере здоровья, инвалидности или смерти;
-
существует и доступно эффективное лечение;
-
начало лечения в доклинический период существенно улучшает прогноз;
-
существует этичный, безопасный и доступный скринирующий тест.
Таким образом, в настоящее время в скрининг можно включать только те генетические нарушения, для которых существует эффективное лечение, но в то же время для заболеваний, не включенных в скрининг, не собирается достаточное количество информации, позволившей бы это лечение разработать. По мнению Дениса Владимировича, требуется сдвиг парадигмы от постнатальной диагностики к преконцепции и предикции — выбору альтернативных методов зачатия ребенка (например, ЭКО) при большом риске развития патологий у ребенка на основе генетического анализа родителей. Также необходимо включение в анализ максимального количества патогенных факторов. Выявление неизлечимых на данный момент пациентов даст возможность перейти к разработке персонализированных препаратов.
Отвечая на вопросы, Денис Владимирович отметил, что разработка персонализированных препаратов бесплатна для родителей и финансируется за счет государственного задания Минздрава. К настоящему времени индивидуальная терапия была разработана для шести пациентов. Ни у кого из них не наблюдались побочные эффекты. У трех зарегистрирован ответ на терапию, у двух, по словам докладчика, понятна причина его отсутствия и ведется дополнительная работа.
На вопрос о соотношении de novo и наследуемых от родителей мутаций, Денис Владимирович ответил, что по их данным соотношение примерно 50 на 50.
Следующий доклад — «Генная терапия лизосомных болезней накопления» — представил Альберт Ризванов, руководитель центра превосходства «Персонифицированная медицина» Института фундаментальной медицины и биологии Казанского федерального университета.
В мире существует более 7000 редких заболеваний, из которых 80% имеют наследственный характер. Около 320 миллионов человек в мире страдают от редких наследственных заболеваний. Лизосомные болезни накопления (ЛБН) — это группа метаболических расстройств, вызванных дефектами в лизосомах — органеллах, ответственных за расщепление и переработку биомолекул. Такие нарушения вызывают накопления непереработанных молекул, что приводит к нарушениям на уровне клеток и органов. К ЛБН относятся болезнь Гоше, болезнь Фабри, мукополисахаридозы и многие другие.
Лаборатория Альберта Анатольевича исследует два таких заболевания. Первое — метахроматическая лейкодистрофия (МЛД) — поражает миелиновые оболочки нейронов. Болезнь возникает из-за дефицита фермента арилсульфатазы А (ARSA) или белка-активатора сапозина B (SapB), участвующих в гидролизе сульфатидов. Регистрируется один случай МЛД на 40 тысяч новорожденных. Симптомы включают гипотонию мышц, нарушения рефлексов, психомоторного и речевого развития, интеллекта, поведения, развитие деменции. Средняя продолжительность жизни больного — около 20 лет после диагноза.
Клинические исследования терапии МЛД включали применение рекомбинантного белка ARSA, которое не дало однозначного улучшения, и трансплантацию гемопоэтических стволовых клеток (ГСК). Последнее позволяло стабилизировать состояние и замедлить развитие заболевания, если проводилось до или на самой ранней стадии развития симптомов.
Участникам клинического исследования, которое в итоге привело к созданию препарата Либмелди/Ленмелди (атидарсаген аутотемцел), вводили внутривенно аутологичные (их собственные) CD34+ ГСК, трансдуцированные лентивирусом LV-ARA. У всех пациентов, которым провели терапию в предсимптомную или раннесимптомную стадию, заболевание не проявлялось или не прогрессировало. Однако состояние пациента с уже развившейся тяжелой двигательной и когнитивной дисфункцией не улучшилось. Атидарсаген аутотемцел зарегистрирован в 2020 году в Евросоюзе, а в 2024 году — в США. Оптовая цена Ленмелди в США — 4,25 млн долларов, что делает его на данный момент самым дорогостоящим препаратом в мире: по цене он опережает даже генотерапевтические препараты для лечения гемофилии В.
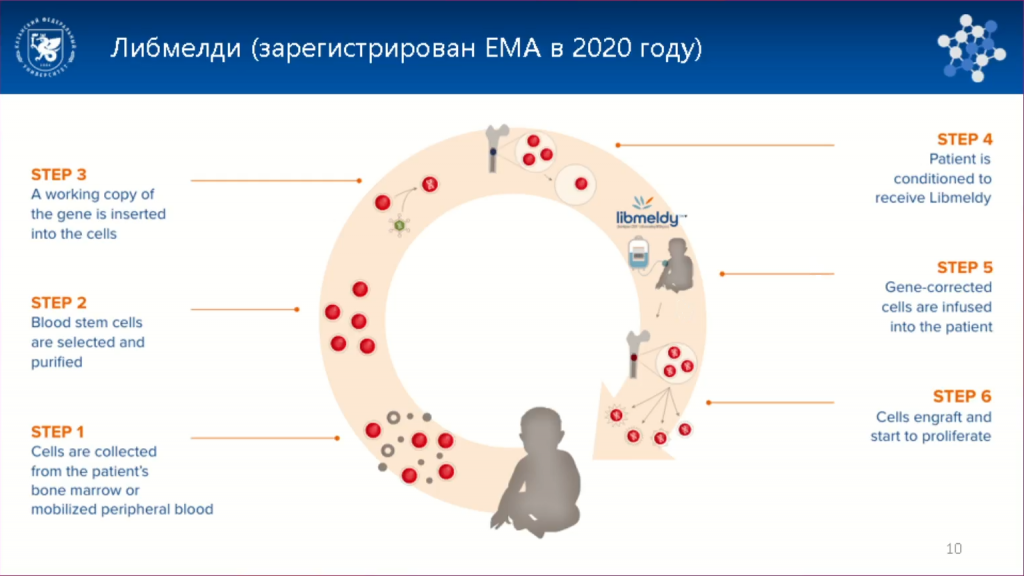
В лаборатории Альберта Анатольевича был создан препарат на основе AAV, несущего ген ARSA. Введение препарата в культуру клеток приводит к высокой активности ARSA. В тестах на нативных (без мутантного гена) свиньях активность ARSA после интратекального и внутривенного введения препарата была показана в различных регионах нервной системы. Присутствие рекомбинантного белка было также подтверждено иммунофлуоресцентным анализом. Патологических процессов не наблюдалось, однако после внутривенного введения в крови животных повысился уровень билирубина. Это свидетельствует о возможном гепатотоксичном действии препарата, которое требует дополнительных исследований.
Второе заболевание, с которым работает лаборатория, GM2 ганглиозидоз, вызвано дефицитом или нарушением функции фермента гексозаминидазы из-за мутаций в гене HEXA (при болезни Тея–Сакса) или HEXB (при болезни Сандхоффа). Симптоматика включает регресс физического и психического развития, потерю зрения, слуха и способности глотать, паралич и атрофию мышц. Частота — примерно один случай на 320 тысяч новорожденных. Заболевание на данный момент неизлечимо, существуют только паллиативные терапии.
В лаборатории докладчика разрабатывались различные методы терапии GM2 ганглиозидоза. Он рассказал об использовании мезенхимных стволовых клеток (МСК), генетически модифицированных с помощью вирусных векторов, несущих гены HEXA и HEXB. Модифицированные клетки культивировали вместе с клетками пациента с болезнью Тея—Сакса в системе трансвелл, где клетки отделены друг от друга полупроницаемой мембраной, пропускающей биомолекулы. При таком культивировании в клетках пациента восстанавливалась активность фермента за счет передачи его от генетически модифицированных МСК. In vivo тесты на крысах (не несущих заболевания) показали повышенную ферментативную активность в ряде тканей, без патологических изменений. Модифицированные МСК обнаруживались в тканях животного гистологическим анализом. Повышенный уровень активности фермента сохранялся в течение месяца, что характерно для подобных тестов на нативных животных.
Завершающий доклад, «Обзор технологии трансфекции в условиях импортозамещения», представила Надежда Гробер, руководитель отдела клеточно-молекулярной биологии ООО «Альгимед».
Компания «Альгимед» предоставляет комплексные решения для оснащения лабораторий, начиная от реагентов и расходных материалов, и заканчивая сложным оборудованием для трансфекции, масс-спектрометрии и пр. Также компания обеспечивает сервисную поддержку и обучение пользователей.
Рынок оборудования для трансфекции, ранее представленный преимущественно американскими и немецкими компаниями, такими как BIORAD, LONZA и Thermo Fisher Scientific, резко сократился после 2022 года. Отечественные компании обратились к китайским поставщикам. В 2023 году была налажено партнерство с китайской компанией Etta Biotech, поставляющей оборудование для трансфекции. Также были найдены поставщики другого оборудования для общелабораторных задач: single cell анализ (MobiDrop), криохранение (Biologix, Laboao), CO2 инкубаторы и сухожары (BOXUN), общелабораторное оборудование (Isolab, Biosan, Laboao, Biobase).
Etta Biotech — первая компания в Китае, которая специализируется на исследованиях и разработках технологии электропорации. На данный момент выпускаются три модели электропораторов: H1 (для базовых задач), F2 (открытого плана с возможностью использования сторонних расходных материалов) и M1 (закрытая система для использования в GNP производстве). В качестве демо-прибора компанией «Альгимед» был оформлен заказ на самую простую модель H1. Надежда Александровна особо отметила преимущество открытой системы данной модели в сравнении с аналогичными приборами LONZA, которые требовали расходных материалов от производителя.

Модель H1 может работать как с планшетами, так и с кюветами, которые можно использовать вплоть до 50 циклов при соблюдении протокола промывки. Etta Biotech имеет собственную технологию электродов HiDEN, которые представляют собой массив игл, создающих напряжение электрического поля. Электроды изготавливаются для 96-, 24- и 12-луночных планшетов, при этом заказчик может использовать собственные планшеты.
Компания «Алкор Био» (Санкт-Петербург) провела анализ эффективности трансфекции на электропораторе X-Porator H1 с помощью проточной цитометрии (в процентах клеток, флуоресцирующих за счет продукции GFP). Эффективность трансфекции достигла 80–94%. Результаты сравнения с нуклеофектором Lonza 2b можно получить по запросу по электронной почте.

По другим моделям приборного ряда данных об эффективности, полученных на территории России, пока не имеется. Согласно данным китайских коллег, модель X-Porator F2 также показывает высокую эффективность.
Для GNP условий Etta Biotech предлагает модель X-Porator M1. Компания рекомендует сначала проводить эксперимент в малом объеме на приборе H1, а затем масштабировать его на H1.
Среди преимуществ расходных материалов для приборов Etta Biotech Надежда Александровна выделила низкую стоимость буфера (в два-три раза дешевле аналогов LONZA); универсальность (нет закрытости систем, кроме модели M1); гибкость (форматы под 12, 24, 96 реакций, возможность использования кювет и планшет) и совместимость материалов с другими брендами для модели F2 (пробирки на 15 и 50 мл Corning). По отзывам пользователей демо-прибора, буфера хватает на продолжительное время.
Связаться с компанией можно по электронной почте cellbio@algimed.ru и по телефону +7 499 391 16 10.
В завершение вебинара Светлана Анатольевна поблагодарила докладчиков и слушателей и пригласила всех присоединяться к каналу портала PCR.NEWS и чату Gene therapy.
Информация о докладчиках
Смирнихина Светлана Анатольевна, к.м.н., доцент, зав. лабораторией редактирования генома ФГБНУ «МГНЦ им. академика Н.П. Бочкова»
Мазунин Илья Олегович, к.б.н., с.н.с., Центр молекулярной и клеточной биологии Сколковского института науки и технологий
Карабельский Александр Владимирович, к.б.н., научный руководитель направления «Генная терапия», Научный центр трансляционной медицины, Университет «Сириус»
Ребриков Денис Владимирович, д.б.н., директор Института трансляционной медицины ФГБУ «НМИЦ АГП им. В.И. Кулакова»
Ризванов Альберт Анатольевич, д. б. н., руководитель центра превосходства «Персонифицированная медицина» Института фундаментальной медицины и биологии Казанского (Приволжского) федерального университета
Гробер Надежда Александровна, руководитель отдела клеточно-молекулярной биологии ООО «Альгимед»



 0
0