Меню
Меню





 Все темы
Все темы
Восемь патогенов, чью эволюционную историю помогла изучить древняя ДНК
Как долго бактерия или вирус циркулирует в человеческой популяции? Всегда ли только человек был хозяином патогена? Протекает ли болезнь, вызванная современными штаммами, так же, как в древности, или иначе? Загадки эволюционной истории патогенов человека помогает разгадывать «симбиоз» находок археологии, успехов в области NGS, которые сделали возможной работу даже с плохо сохранившейся древней ДНК, и методов молекулярного датирования на основе байесовского подхода.

Пляска смерти. Нигулисте, Таллин. XV в. | Wikimedia.org
Как измерить возраст патогена?
Можно ли измерить время эволюционного расхождения двух видов числом мутаций в их ДНК? Впервые эта идея возникла у Эмиля Цукеркандля и Лайнуса Полинга еще в 1962 году — правда, тогда они думали о мутациях в белковых молекулах. Сегодня такой подход вполне реален, и существуют методы молекулярного датирования с помощью филогенетических деревьев.
Различие в нуклеотидных последовательностях организмов само по себе может обеспечить только относительную оценку, например, можно определить, что два вида А и В разошлись друг с другом позднее, чем вид С с общим предком А и В. Для пересчета относительного времени расхождения в абсолютное необходима калибровка временной шкалы. Для этого информацию о последовательностях дополняют данными о возрасте образцов, известном из независимых источников (подробнее о филогенетических деревьях, общих предках и молекулярных часах можно почитать в этом материале).
Основная цель молекулярного датирования — преобразовать различие между последовательностями ДНК в абсолютный возраст внутренних узлов филогенетического дерева, то есть определить предполагаемое время до момента существования самого последнего общего предка интересующих нас организмов (Time to the Most Recent Common Ancestor, TMRCA). Внутренние узлы как раз и представляют собой таких предполагаемых предков для всех отобранных особей анализируемой группы. Некоторые из узлов соответствуют ключевым событиям в эволюционной истории вида.
Как древняя ДНК помогает узнать эволюционный возраст?
В одной из стратегий калибровки молекулярных часов используется информация о возрасте самих секвенированных образцов (в случае патогенов – год выделения изолята). Однако в этом случае точное датирование возможно, только если разброс в возрасте изучаемых образцов и, соответственно, число замен достаточно велики. Лучше всего такая стратегия подходит для быстроразвивающихся таксонов (например, РНК-содержащих вирусов), если образцы отобраны серией, а в других случаях как раз будут полезны данные древней ДНК.
Так, в случае недавно появившихся зоонозов можно ответить на многие вопросы об их эволюции, используя только современные образцы. Это касается, например, вирусов гриппа, коронавирусов или ретровирусов (к последним относится ВИЧ). Они эволюционируют достаточно быстро, и их можно изучать в режиме реального времени. Например, мы хорошо помним, как ученые сравнивали последовательности изолятов различных штаммов коронавируса, полученные в разных странах, и делали выводы о том, когда и где мог возникнуть тот или иной штамм.
Другие патогены эволюционируют медленно и мутируют почти с той же скоростью, что и сам геном человека. У патогенов с двухцепочечной ДНК (например, герпесвирусов) скорость замен нуклеотидов настолько низкая, что даже образцов за 50 лет может быть недостаточно для адекватной калибровки молекулярных часов — необходимы геномы древних вирусов.
В изучении древней ДНК (ancient DNA, аДНК) на протяжении последнего десятилетия произошел прорыв. Ученым удалось восстановить многочисленные полногеномные последовательности из глубокого прошлого. Этот прорыв сделала возможным огромная производительность современных технологий секвенирования, которые позволяют проанализировать даже короткие и деградированные молекулы ДНК. А самое главное — анализ миллиардов считываний для каждого образца обеспечивает количественную оценку уровня контаминации (примеси современной и/или посторонней ДНК) — решение проблемы, которая десятилетиями осложняла работу исследователям аДНК.
Здесь мы расскажем о патогенах, эволюционную историю которых позволило уточнить добавление последовательностей аДНК в выборку для анализа. Рассмотрим их от самого древнего к самому молодому, считая по времени возникновения ветвей, включающих современные варианты.
Парвовирус B19, возбудитель инфекционной эритемы: «Я старше, чем вы могли представить»
Современные варианты появились примерно 7190 лет назад.
О патогене
Парвовирусы — небольшие безоболочечные вирусы с одноцепочечной ДНК (оцДНК) и размером генома около 5 килобаз. Вирус B19 реплицируется в костном мозге, а затем проникает в другие ткани, прежде всего в легкие, и передается через респираторные секреты. Заболевание у здоровых детей и взрослых обычно протекает в легкой форме и проходит самостоятельно. У маленьких детей наиболее частое проявление первичной инфекции — инфекционная эритема (пятая болезнь, синдром пощечины) — сыпь на лице интенсивно красного цвета. Однако при внутриутробном заражении в 2–10% случаев инфекция матери приводит к летальному исходу для плода.
Классификация
В настоящее время ученые выделили три генотипа парвовируса B19: генотип 1 (распространен во всем мире и преобладает в Европе), генотип 2 (редкий в Европе, обычно обнаруживается только у людей, родившихся до середины 1970-х годов) и генотип 3 (наиболее распространен в Африке).
Первоначальная гипотеза о времени происхождения
Кто исследовал
Ученые из Великобритании в 2007 году.
Какие образцы изучали
Нуклеотидные последовательности современных образцов из Ганы, Европы и Бразилии.
Возраст патогена
Скорость эволюции рассчитана как одна из самых высоких среди вирусов с оцДНК — в диапазоне 1–2 × 10 -4 нуклеотидных замен на сайт в год, что сопоставимо с некоторыми РНК-содержащими вирусами. Согласно этим расчетам, общий предок существующих вариантов B19 появился относительно недавно: в случае генотипа 3 всего около 500 лет назад, а у генотипа 1 не ранее, чем в 1950-е годы.
Но эти данные противоречат наблюдаемому разнообразию циркулирующих в мире вариантов парвовируса B19.
Что помогла выяснить древняя ДНК
- Возраст самого вируса
Кто исследовал
Международная команда ученых, в которую вошли сотрудники Зоологического института РАН (Санкт-Петербург) и Иркутского государственного университета в 2018 году.
Какие образцы изучали
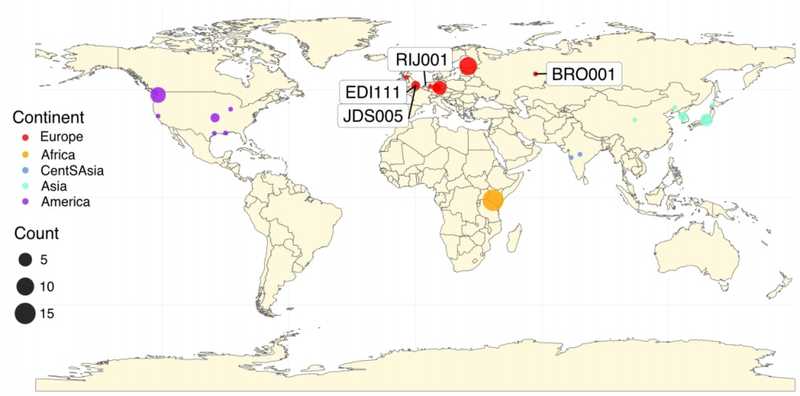
Десять образцов древней ДНК из человеческих останков и все имеющиеся полные геномы из современных образцов, собранных при первичной инфекции и персистенции вируса у взрослых разного возраста. Источниками аДНК вируса B19 были зубы, костный мозг и каменистая часть височной кости людей, живших в Европе, Центральной Азии и Гренландии. Образцы возрастом от 500 до 6900 лет относились к разным археологическим культурам: четыре скандинава эпохи викингов, три байкальских охотника-собирателя эпохи раннего неолита и раннего бронзового века, один ранний славянин из Чехии и один представитель народа гуннов из киргизского Тянь-Шаня. Самый древний образец был найден на территории России около Байкала.
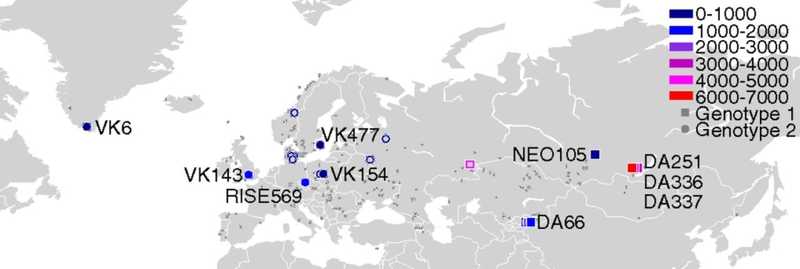 Места обнаружения образцов, содержащих геномы парвовируса B19. Образцы отмечены разными цветами в соответствии с возрастом. Квадраты обозначают генотип 1, кружки — генотип 2 (Mühlemann B., 2018)
Места обнаружения образцов, содержащих геномы парвовируса B19. Образцы отмечены разными цветами в соответствии с возрастом. Квадраты обозначают генотип 1, кружки — генотип 2 (Mühlemann B., 2018)
Уточненный возраст патогена
На построенном учеными филогенетическом дереве пять древних образцов вошли в состав генотипа 1 или были предковыми для него (синий цвет на дереве), а пять образцов — предковыми для генотипа 2 (красный цвет на дереве). Вновь рассчитанная скорость эволюции парвовируса B19V оказалась на порядок ниже, чем предполагалось ранее, — 1,22 × 10 −5 замен на сайт в год. Это означает, что общий предок современных линий вируса появился в популяции человека не около 1800 года, а, вероятно, около 8000 года до н.э. Последние общие предки (MRCA) генотипов 1, 2 и 3 циркулировали около 7, 2 и 2,5 тысячи лет назад соответственно.
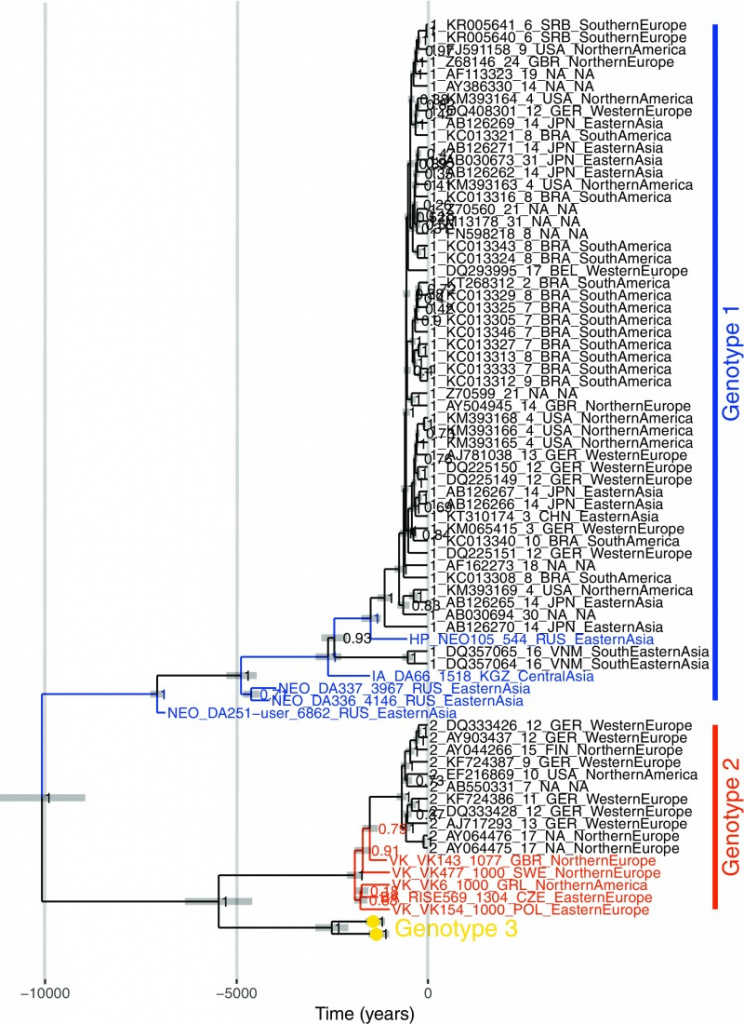 Филогенетическое дерево, включающее современные и древние геномы парвовируса В19 (Mühlemann B., 2018)
Филогенетическое дерево, включающее современные и древние геномы парвовируса В19 (Mühlemann B., 2018)
- Возраст генотипа 3
Кто исследовал
Ученые из Мексики, Дании и США в 2021 году.
Какие образцы изучали
26 образцов зубов людей, предположительно имевших африканское происхождение. Останки были найдены в массовых захоронениях жертв эпидемии («коколицтли», 1576 год) возле больницы и часовни на территории нынешнего Мехико. Удалось восстановить три генома древнего парвовируса B19, которые были отнесены к генотипу 3.
Уточненный возраст патогена
Восстановленные геномы были родственны африканским штаммам. Это позволяет предположить, что вирус возник на африканском континенте и был занесен в Мексику в период работорговли. Скорость эволюции парвовируса составила от 1,03× 10−5 до 2,62× 10−5 замен/сайт/год в зависимости от выбранных параметров. MRCA генотипов 1, 2 и 3 циркулировали 7,19, 2,11 и 3,64 тысяч лет назад, соответственно, а их общий предок — 11,5 тысяч лет назад.
Вирус простого герпеса 1 типа (ВПГ-1): «Я весь такой противоречивый»
Современные варианты появились примерно 5230 лет назад (но это не точно).
О патогене
ВПГ-1 — вирус с двухцепочечной ДНК, встречается как минимум у двух третей взрослого населения в мире. Инфицирование обычно происходит в младенчестве или детстве, протекает в легкой форме или бессимптомно. После первичного заражения вирус становится латентным и персистирует в сенсорных нейронах. При психологическом или физиологическом стрессе ВПГ-1 может пробуждаться, вызывая рецидивирующие поражения губ. Иммунная система здорового человека обычно предотвращает значительную виремию.
Классификация
Современные ВПГ-1 можно разделить на три филогруппы, связанные с их географическим происхождением: I (Европа и Америка), II (Европа, Азия и Америка) и III (Африка).
Первоначальная гипотеза о времени происхождения
Кто исследовал
Ученые из Италии в 2020 году.
Образцы
140 современных геномов ВПГ-1 из в баз данных ViPR и NCBI. Самый ранний образец — штамм от взрослого человека из США, выделенный в 1980 году.
Возраст патогена
Расчеты показали достаточно позднее происхождение ВПГ-1, а источник его распространения был в Африке. Циркуляция общего предка всех штаммов вируса датирована около 7000 лет назад, а расхождение базальной клады, содержащей в основном африканские последовательности, и всех других штаммов произошло примерно 5000 лет назад.
Что помогла выяснить древняя ДНК
Кто исследовал
Команда специалистов из Эстонии, Великобритании, Германии, Италии и России в 2022 году (подробнее на PCR.NEWS).
Образцы
Древние геномы ВПГ-1, обнаруженные в зубах четырех человек из археологических памятников Северной Европы. Это были мужчина с городского средневекового больничного кладбища (1350–1450 гг.) и женщина с кладбища раннего англосаксонского периода в графстве Кембриджшир (500–575 гг.) в Великобритании, мужчина из погребения неволинской культуры (253–530 гг.) в России и мужчина из Нидерландов (1600–1700 гг.).
Уточненный возраст патогена
Скорость эволюции ВПГ-1 при разных моделях составила от 2,38 × 10 −7 до 2,49 × 10−6 замен на сайт в год. MRCA евразийских штаммов с наибольшей вероятностью циркулировал 4,68 тыс. лет назад. При этом MRCA европейской филогруппы I и паневразийской филогруппы II присутствовали 4,50 и 4,47 тыс. лет назад, а от африканской филогруппы III они отделились около 5,29 тыс. лет назад. Это вполне совпадает с оценками на основе современных последовательностей вируса. Авторы предположили, что распространение филогрупп I и II и замещение ими филогруппы III совпало с периодом позднего неолита и последовало за миграцией людей бронзового века из Африки. Авторы предположили, что в процессе замены более старых штаммов современными кроме изменений в популяционной динамике сыграло роль появление новой культурной практики — эротического поцелуя. Это увеличило количество оральных контактов и могло способствовать горизонтальной передаче, характерной для современной эпидемиологии вируса. Впрочем, данное предположение остается дискуссионным.
 Места обнаружения образцов, содержащих современные и древние геномы ВПГ-1 (Guellil M, 2022)
Места обнаружения образцов, содержащих современные и древние геномы ВПГ-1 (Guellil M, 2022)
К сожалению, в этом случае древняя ДНК не помогла ответить на все вопросы. Возраст проанализированных вирусов все-таки не позволяет изучить, как вирус эволюционировал до их циркуляции. По расчетам, проведенным в 2013 году на основе выборки из 31 полного генома современных изолятов ВПГ-1 и одного генома ВПГ-2, расхождение этих двух вирусов произошло около 2,18 миллиона лет назад, при этом ВПГ-1 начал распространяться примерно 50 300 лет назад. Фрагменты ДНК ВПГ-1 были обнаружены в молочных зубах двух детей, живших более 30 тысяч лет назад. Качество ДНК было недостаточным для реконструкции генома плейстоценового ВПГ-1, но это подтверждает древность патогена и оставляет простор для дальнейших исследований.
Mycobacterium tuberculosis, возбудитель туберкулеза: «Я моложе, чем вы рассчитывали»
Современные варианты появились примерно 3258 лет назад.
О патогене
Возбудители туберкулеза —- бактерии комплекса Mycobacterium tuberculosis (MTBC). К нему относятся M. tuberculosis, M. bovis, M. canettii, M. africanum, M. pinnipedii, M. microti, M. caprae, М. mungi и М. orygis. Чаще всего туберкулез у человека вызывают первые два микроба. Микобактерии обычно поражают легкие, реже затрагивая другие органы и системы. Передаются воздушно-капельным путем. После инфицирования болезнь обычно протекает в скрытой форме, но примерно один из десяти случаев инфекции переходит в активную форму.
Классификация
В пределах комплекса MTBC можно выделить семь филогенетических линий, связанных с различными регионами мира: линия 1 (Восточная Африка, Филиппины); современные евроазиатские линии 2, 3 и 4, которые находятся в тесном филогенетическом родстве; африканские линии 5 и 6, включающие M. africanum и распространенные в Западной Африке; линия 7, обнаруженная у больных туберкулезом из Эфиопии.
Хронологическое положение линий определяется наличием или отсутствием MTB-специфической делеции (TbD1): у предковых она есть (TbD1+), а у современных нет (TbD1-). На этом основании линии 1, 5, 6 и 7, линии животных и M. canettii отнесены к предковой группе MTBC, а линии 2, 3 и 4 — к современной с эволюционной точки зрения. Эти данные позволили предположить, что линии микобактерий животных и M.africanum разошлись от прародителя предковых линий MTBC и что M. tuberculosis изначально была человеческим патогеном.
Первоначальная гипотеза о времени происхождения
Кто исследовал
Международная команда ученых в 2013 году. Они выдвинули гипотезу о том, что разные линии MTBC могут быть связаны с гаплогруппами митохондриальной ДНК человека (N, M и L).
Какие образцы изучали
Полные геномы 259 современных клинических изолятов микобактерий, а также глобальный датасет мтДНК человека (n = 4955), датасет мтДНК времен неолитической экспансии (n = 423) и датасет, дополненный данными мтДНК из Восточной Азии времен неолита (n = 72). Для калибровки филогении MTBC использовали временны́е точки человеческой эволюции (появление современного человека 85 тысяч лет назад, возникновение митохондриальной гаплогруппы L3 — 70 тысяч лет назад, и др.).
Возраст патогена
MTBC появился около 70 тысяч лет назад в Африке и сопровождал миграцию Homo sapiens из этой области. В эпоху неолита генетическое разнообразие MTBC существенно расширилось из-за увеличения плотности населения.
Авторы предположили, что разные линии адаптировались к разным гаплогруппам людей. Топология ветвления митохондриальных макрогаплогрупп M и N, происходящих из Африки, от исходной африканской гаплогруппы L3 отражала филогению MTBC — связи между линией 1, евразийскими линиями 2/3/4 и африканскими линиями 5 и 6 были очень схожи с деревом человеческих гаплогрупп.
По мнению ученых, развиваясь параллельно со своим хозяином-человеком, M. africanum, M. canettii и линия 7 остались в Африке, тогда как другие линии могли достичь Европы и Азии после колонизации этих территорий. Была обнаружена корреляция между дивергенцией линии 1 около 67 тысяч лет назад и первой волной миграции человека из Африки в район Индийского океана. Другой раскол MTBC, который мог произойти 46 тысяч лет назад, привел к появлению линий 2, 3 и 4; по времени он соответствует второй волне миграции Homo sapiens из Африки в Евразию.
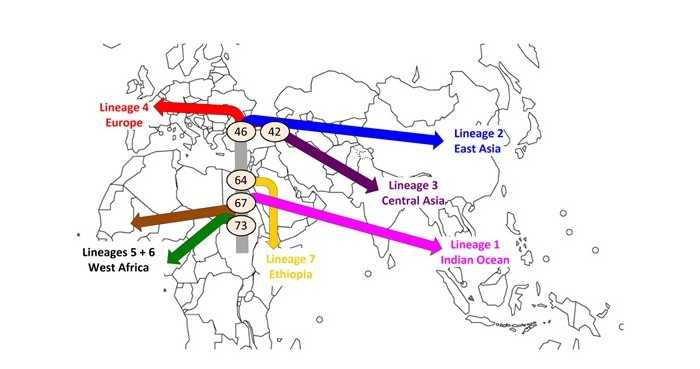 Карта, суммирующая результаты филогеографического и датировочного анализа MTBC. Разные линии отмечены разными цветами. Числами обозначено время расхождения линий в тыс. лет назад (Comas I, 2013)
Карта, суммирующая результаты филогеографического и датировочного анализа MTBC. Разные линии отмечены разными цветами. Числами обозначено время расхождения линий в тыс. лет назад (Comas I, 2013)
Что помогла выяснить древняя ДНК
Кто исследовал
Ученые из Германии, Дании и Швеции в 2020 году.
Образцы
ДНК выделили из кальцинированного узла в правом легком Педера Винструпа, епископа шведского города Лунда (1605–1679). Тело епископа, захороненное в саркофаге и естественным образом мумифицированное, начали исследовать в 2015 году (подробнее о епископе на PCR.NEWS). Удалось получить геномные данные древней M. tuberculosis самого высокого качества. Также использовался набор данных, отражающий современное разнообразие MTBC, и шесть древних геномов микобактерий в качестве калибровочных точек. Помимо «штамма епископа», это были два штамма M. tuberculosis из венгерских мумий конца XVIII и начала XIX веков и три штамма M. pinnipedii из человеческих останков возрастом около 1000 лет, найденных на территории Перу. В качестве внешней группы для моделирования времени отделения современных линий MTBC от общего предка ученые взяли геном M. canettii.
Уточненный возраст патогена
По оценке авторов, микобактерии возникли в интервале между 2000–6000 годами до настоящего времени. Время циркуляции MRCA датировали приблизительно 1248 годом до н.э. (около 3258 лет назад). То есть использование данных древней ДНК в качестве калибровочных точек показало более поздние даты происхождения MTBC.
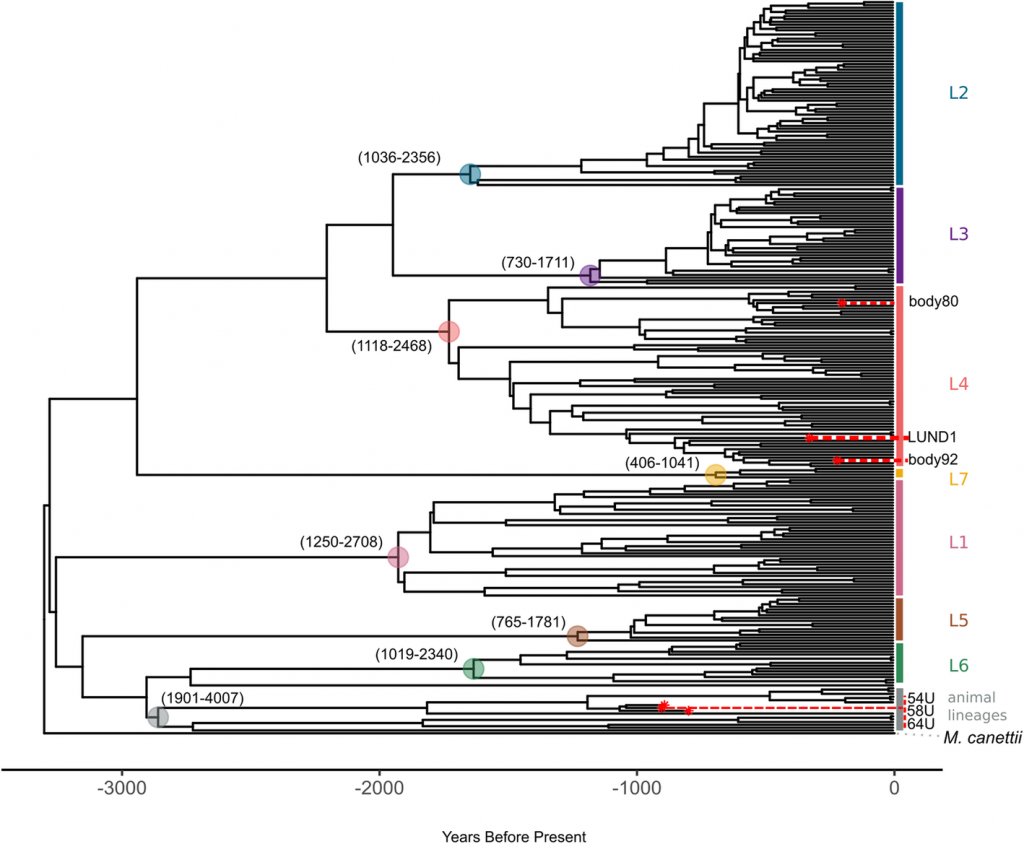 Филогенетическое дерево на основе полного набора данных MTBC. Линии отмечены справа. Древние геномы обозначены красными звездочками, а сбоку отмечены их названия. Предковые узлы выделены кружком, в скобках указан доверительный интервал. Временная шкала выражена в годах до настоящего времени (самый поздний образец выделен в 2010 году) (Sabin, S., 2020)
Филогенетическое дерево на основе полного набора данных MTBC. Линии отмечены справа. Древние геномы обозначены красными звездочками, а сбоку отмечены их названия. Предковые узлы выделены кружком, в скобках указан доверительный интервал. Временная шкала выражена в годах до настоящего времени (самый поздний образец выделен в 2010 году) (Sabin, S., 2020)
Mycobacterium leprae, возбудитель проказы: «Утратил былое разнообразие»
Современные варианты появились 2300–2890 лет назад.
О патогене
Проказа (болезнь Хансена) — одно из старейших заболеваний, известных человечеству. Самые ранние описания предполагаемой проказы содержатся в египетских папирусах (1550 г. до н. э.) и в индийском Сушрута-самхите (600 г. до н. э.). Более достоверные сведения о болезни Хансена можно найти в древнегреческой и римской литературе начиная с первого века нашей эры. Проявления проказы включают повреждение периферической нервной системы, слизистых оболочек, кожи и в итоге конечностей.
Болезнь Хансена была широко распространена в Европе в средние века. Для размещения и изоляции заболевших людей строили лепрозории. В отличие от чумы, требовавшей экстренных захоронений, это медленно развивающееся заболевание позволяло мирно похоронить умерших на кладбищах при лепрозории.
Классификация
При исследовании филогенетического разнообразия геномы M. leprae были сгруппированы в шесть основных ветвей (0–5), а далее ветвь 2 была разделена на три подветви (2E, 2H и 2F). Почти все основные ветви, в том числе самая базальная (ветвь 0), которая включает и современные восточноазиатские образцы, присутствовали и в средневековой Европе.
Первоначальная гипотеза о происхождении
Наиболее вероятным местом происхождения патогена считается Индия. Предполагается, что он проник на запад во время завоеваний Александра Великого (IV до н.э.) или посредством торговли, а впоследствии с расширением Римской империи распространился по Средиземноморскому бассейну и в Западную Европу (200 г. до н.э. — 600 г. н.э.). Анализ археологических находок скелетов людей с болезнью Хансена подтвердил эту версию. До недавнего времени самые древние останки людей с признаками инфекции из Индии были датированы II тысячелетием до нашей эры, из Италии — IV-III веками до нашей эры, из Египта — III веком до нашей эры, из Израиля — I веком нашей эры. Однако также были обнаружены генетически не подтвержденные случаи из Венгрии и Шотландии бронзового века, которые ставили под сомнение гипотезу азиатского происхождения проказы.
Что помогла выяснить древняя ДНК
Кто исследовал
Международный коллектив ученых, в состав которого вошли специалисты из Москвы и Казани.
Какие образцы изучали
Древнюю ДНК от 41 человека, в том числе 39 с остеологическими или историческими доказательствами болезни Хансена и двух положительных на M. leprae при генетическом исследовании. Останки датируются VI-XX веками нашей эры и обнаружены в 20 археологических памятниках по всей Европе, включая территории, для которых ранее не существовало полногеномных данных (Беларусь, Иберия, Россия и Шотландия). Для оценки разнообразия штаммов внутри регионов ученые исследовали скелеты из нескольких локаций в Кембриджшире (Англия) и останки людей, похороненных в двух лепрозориях в Португалии и Испании. Всего удалось реконструировать 19 геномов M. leprae, которые были пригодны для филогенетического анализа, из них 16 геномов имели достаточное качество для молекулярного датирования в BEAST. Ученые объединили эти образцы со 177 уже известными современными и древними геномами M. leprae.
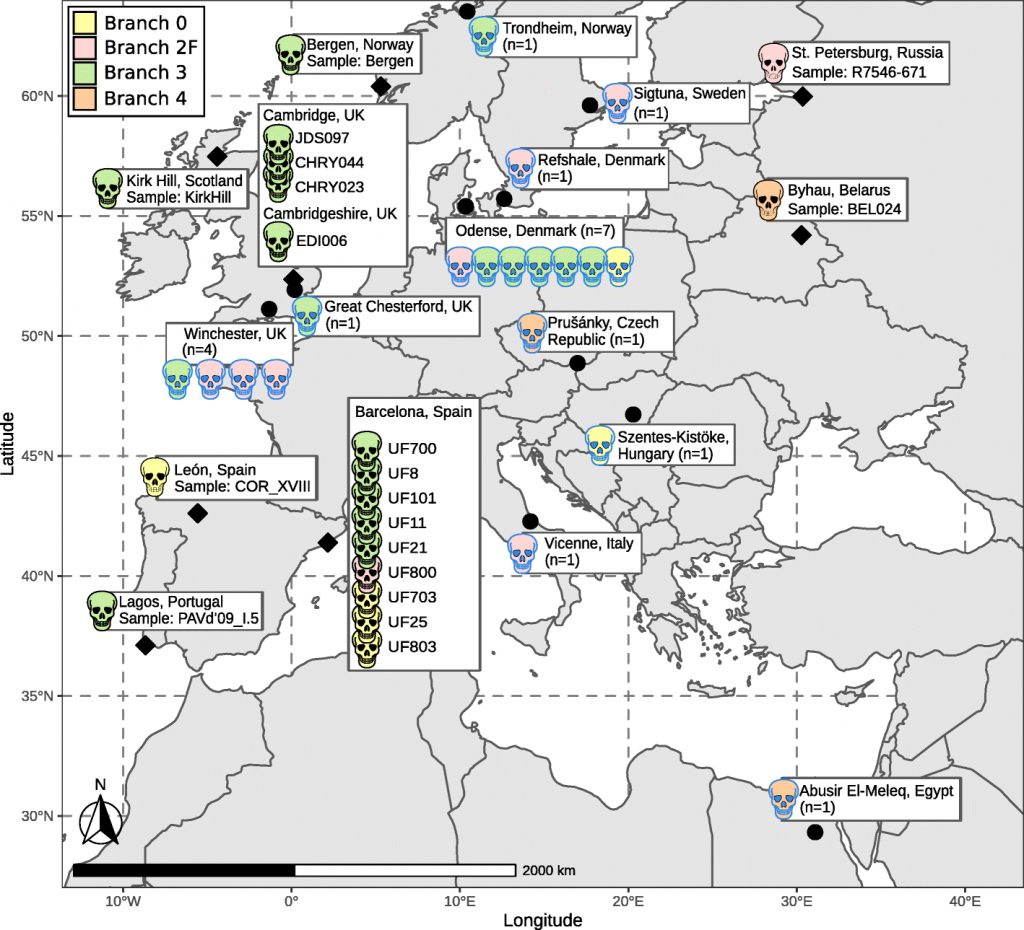 Места обнаружения образцов, из которых были реконструированы древние геномы M. leprae. Цвет черепов соответствует ветвям филогении M. leprae. Ромбы указывают расположение объектов, охваченных исследованием Pfrengle S et al.; кружки и черепа с синим контуром показывают места обнаружения штаммов, опубликованных в более ранней работе Schuenemann VJ et al.
Места обнаружения образцов, из которых были реконструированы древние геномы M. leprae. Цвет черепов соответствует ветвям филогении M. leprae. Ромбы указывают расположение объектов, охваченных исследованием Pfrengle S et al.; кружки и черепа с синим контуром показывают места обнаружения штаммов, опубликованных в более ранней работе Schuenemann VJ et al.
Уточненный возраст патогена
Ближайший общий предок всех последовательностей, включенных в анализ, циркулировал около 1900 года до н.э. Общие предки отдельных линий, включающих современные штаммы от людей, присутствовали в популяции в период между 870–280 гг. до н.э. (примерно 2300– 2890 лет назад).
Большинство средневековых геномов M. leprae вошло в ветвь 3 и было расположено базально по отношению к современным штаммам возбудителя. Образец из России и другие средневековые европейские последовательности были расположены в ветви 2F. Геном из Беларуси вошел в состав ветви 4, базально по отношению к большинству других геномов внутри нее, включая современные. Четыре иберийских генома относились к ветви 0 — этот кластер является предковым для современных последовательностей генома возбудителя проказы человека и образует сестринскую кладу для штаммов, выделенных от современных обезьян. Также было обнаружено высокое родство двух средневековых геномов из Барселоны и штаммов, выделенных от белок.
Ученые отметили два эпизода значительного расширения популяции возбудителя, которые совпадают с важными периодами в истории человека: диапазон 250 г. н.э. ± 250 лет охватывает период римских завоеваний, а более поздний диапазон, 1600 н.э. ± 150 лет, соответствует быстрому развитию знаний и технологий в позднем Средневековье, кульминацией которого стало начало регулярной трансатлантической торговли.
Отсутствие более древних образцов не позволило определить место происхождения болезни Хансена. Единственный за пределами Европы геном из Древнего Египта расположен базально по отношению к ветви 4 - он не может помочь прояснить происхождение других ветвей, которые присутствовали в средневековой Европе. Интересно, что сегодня регионы со значительной распространенностью болезни Хансена имеют более низкие уровни разнообразия M. leprae, чем штаммы, обнаруженные в средневековых лепрозориях, и в целом в пределах средневекового европейского континента.
Yersinia pestis, возбудитель чумы: «В любой непонятной ситуации эволюционируй!»
Современные варианты появились примерно 683–707 лет назад
О патогене
Yersinia pestis — грамотрицательная, неспорообразующая и неподвижная коккобацилла. По своему антигенному составу и клеточным структурам она сходна с другими кишечными бактериями и продуцирует короткоцепочечный шероховатый липополисахарид, в котором отсутствует О-антиген из-за мутаций некоторых генов. Эта особенность способствует системному заражению и могла быть обусловлена адаптивной эволюцией, превратившей Y. pestis в смертельный патоген.
Y. pestis вызвала три пандемии в истории: первая — Юстинианова чума (примерно с 541 года н. э.), вторая — Черная смерть (примерно с 1347 года н. э.), третья — современная чума (примерно с 1880 года н. э.). В настоящее время Y. pestis встречаются на всех континентах, кроме Антарктиды; ее природные очаги распространены в Азии, Африке и Америке и охватывают влажные и засушливые регионы, луга, пустыни, плато и равнины.
Классификация
В пределах Y. pestis выделили три биовара (Antiqua, Medievalis и Orientalis), которые образовали отдельные ветви филогенетического дерева. Биовар Antiqua вызвал пандемию чумы в VI веке, Medievalis стал причиной Черной смерти и последующих эпидемий во время второй волны пандемии, а Orientalis вызвал современную пандемию чумы. В филогении современных штаммов патогена выделяют пять основных ветвей, которые обозначены как 0, 1, 2, 3 и 4.
Первоначальная гипотеза о времени происхождения
Кто исследовал
Ученые из Германии и Франции в 1999 году.
Какие образцы изучали
Секвенировали фрагменты пяти генов домашнего хозяйства и гена, участвующего в синтезе липополисахарида, для 36 штаммов, представляющих глобальное разнообразие Y.pestis, а также 12 и 13 штаммов двух других патогенных видов иерсиний (Y. pseudotuberculosis и Y. enterocolitica). Штаммы Y. pestis были выделены в период с 1942 по 1998 гг. от человека, блох и мелких млекопитающих в разных странах.
Происхождение патогена
Ученые не обнаружили существенного разнообразия аллелей ни в одном гене Y. pestis; они были идентичны или почти идентичны аллелям Y. pseudotuberculosis. Был сделан вывод, что Y. pestis представляет собой клон, который произошел от Y. pseudotuberculosis 1500–20 000 лет назад, незадолго до первых известных пандемий чумы.
Что помогла уточнить древняя ДНК
- Происхождение чумной палочки
Кто исследовал
Ученые из Германии и Латвии в 2021 году.
Какие образцы изучали
Образец каменного века, обнаруженный в Риннукалнс на севере Латвии, — останки охотника-собирателя 20–30 лет, жившего 5300–5050 лет назад. В выборку для анализа были включены 278 геномов, из которых 276 — ранее опубликованные древние и современные геномы Y. pestis и один — геном Y. pseudotuberculosis.
Уточненный возраст патогена
Y. pestis дивергировала от Y. pseudotuberculosis около 7000 лет назад, в начале неолита. В дальнейшем предковые штаммы Y. pestis распространились по всей поздненеолитической Евразии. В самых ранних линиях патогена отсутствовали генетические адаптации, необходимые для эффективной передачи через блох. Два самых древних генома классифицируют как отдельную линию, существовавшую до позднего неолита — раннего бронзового века. Другая, более поздняя линия включает уже 26 штаммов, которые обнаружены на обширной территории от озера Байкал до Центральной Европы. До своего исчезновения они циркулировали не менее 2500 лет. Предположительно первоначальная форма чумы была легочной.
В бронзовом веке появились и древние линии, имеющие все генетические адаптации для высокоэффективной формы передачи через блох. Самый старый геном с полным набором таких генетических признаков и способный вызывать бубонную чуму (RT5), обнаружен в образце из Самарской области (Россия).
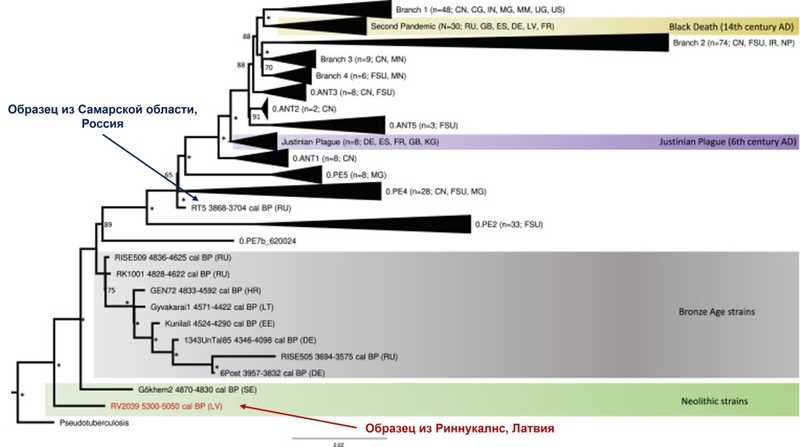 Филогенетический анализ и молекулярное датирование современных и древних штаммов Y pestis (Susat J, 2021)
Филогенетический анализ и молекулярное датирование современных и древних штаммов Y pestis (Susat J, 2021)
- Происхождение современных штаммов
Кто исследовал
Специалисты из международной группы, в которую вошли также ученые из Москвы и Санкт-Петербурга, в 2022 году.
Какие образцы изучали
Геномы Y. pestis, полученные от трех человек, которые были эксгумированы на кладбище Кара-Джигач, расположенного недалеко от озера Иссык-Куль на территории Кыргызстана. На этом кладбище обнаружено непропорционально большое количество захоронений, датируемых 1338–1339 годами, причем в некоторых надгробных надписях указано, что причиной смерти стала неустановленная моровая язва (подробнее на PCR.NEWS). Два из трех геномов были идентичны, что согласуется с опубликованными данными, показывающими низкое разнообразие геномов Y. pestis, изолированных в условиях одной эпидемии. Для последующего анализа данные этих двух образцов были объединены. Всего в выборку вошли 203 современных и 46 исторических геномов Y. pestis.
Уточненный возраст патогена
Расположение образца из Кара-Джигач было предковым для линий 1-4, включающих все известные геномы четырнадцатого века из Западной Евразии и современные штаммы. Опубликованные геномы Второй пандемии были связаны с ветвью 1. Исследователи оценили время разделения ветвей 1–4 в интервале от 1316 до 1340 гг. н.э.
Традиционно начало Черной смерти связывают со вспышками, произошедшими в Причерноморье в 1346 году. Эпидемия в Кара-Джигач началась за восемь лет до этого момента. Основываясь на исторических данных и артефактах, в том числе надписях на надгробиях и кладах монет, авторы предположили, что вблизи Иссык-Куля и реки Чу было перекрестье торговых путей. Там селились представители разных народов и жили за счет торговли с разными регионами Евразии. Это могло способствовать распространению чумы в XIV веке.
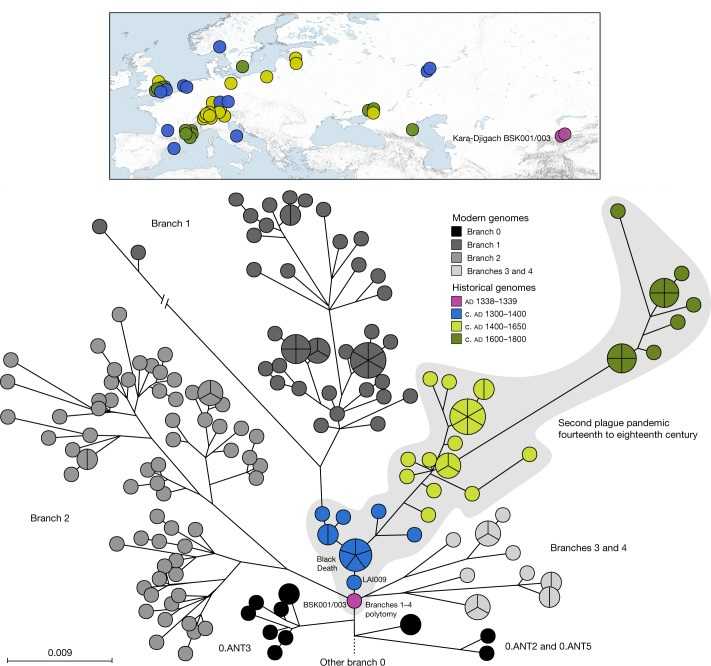 Генетическое разнообразие и места обнаружения древних образцов Y. pestis (Spyrou MA, 2022)
Генетическое разнообразие и места обнаружения древних образцов Y. pestis (Spyrou MA, 2022)За глобальное распространение патогена на чумных кораблях во время третьей пандемии в конце XIX века ответственна самая молодая группа в ветви 1 (1.ORI). Всего по миру прокатились три волны: первая достигла США, вторая в виде множественных заносов - Европы, Южной Америки, Африки и Юго-Восточной Азии, а третья - Мадагаскара и Турции. Молекулярное датирование, проведенное учеными из США, Канады и Австралии, показывает, что общий предок группы 1.ORI циркулировал в период между 1806 и 1901 гг. (подробнее на PCR.NEWS). Это хорошо согласуется с известными эпидемиологическими данными – первые очаги появились в Южном Китае в 1772–1880 гг., а затем чума распространилась по всему миру из Гонконга в 1894–1901 гг.
Вирус оспы: «Поражал викингов, а в ХХ веке сам побежден»
«Современные» варианты появились примерно 332 года назад (а потом вымерли).
О патогене
Ортопоксвирусы, к которым относится вирус оспы (VARV), имеют длинные линейные двухцепочечные ДНК-геномы. По оценкам, оспа была самым смертоносным инфекционным заболеванием в истории человечества из-за совокупного числа смертей, связанных с инфекцией и ее осложнениями. В то же время вирус оспы — единственный патоген человека, уничтоженный с помощью вакцинации, окончание которой было объявлено в 1977 году.
Во время острой фазы заболевания у большинства больных на коже появлялись характерные наполненные жидкостью волдыри, которые у тех, кто пережил первоначальное заражение, в итоге отпадали, часто оставляя обширные рубцы. Высушенные струпья или образцы кожи с волдырями сохранились в исторических и медицинских коллекциях, что является как видимым свидетельством оспы, так и потенциальным источником вирусной ДНК. Поскольку вирус оспы вымер, лабораторные образцы из коллекций в США и в России, исторические образцы, такие как инфицированная высушенная кожа и древняя ДНК из костей, — единственные источники данных для изучения генетической эволюции VARV.
Классификация
Большинство известных геномов вируса относятся к периоду Интенсифицированной программы ВОЗ по ликвидации оспы (1967–1980 гг.) — кампании по ликвидации заболевания в Африке, Азии и Южной Америке и предотвращению распространения оспы в Европу и другие регионы. Основываясь на этих геномах, ученые подразделяют разнообразие вируса на две линии: основная P-I, включающая изоляты из Европы, Азии и Африки, выделенные с 1947 по 1975 гг., и P-II — из Африки, Бразилии и Великобритании периода 1952-1969 гг.
Первоначальная гипотеза о времени происхождении
Кто исследовал
Две международные команды ученых в 2016 и 2020 годах.
Какие образцы изучали
Исторический — мумию ребенка, датированную 1654 годом (Вильнюс, Литва) и музейный — зафиксированную этанолом ногу младенца, залитую в жидкий парафин, препарированную знаменитым шотландским хирургом Джоном Хантером между 1760 и 1793 годами (Хантеровский музей Королевского колледжа хирургов, Англия).
Возраст патогена
Исследования показали относительно молодой возраст вируса оспы. Скорость эволюции была оценена в диапазоне 8,5 × 10 −6 — 10,67 × 10 −6 замен на сайт в год. Это позволило предположить, что изоляты VARV, отобранные в период с 1944 по 1977 год, имели относительно недавнего общего предка, сформировавшегося в XVI–XVII вв., возможно, в результате прохождения популяции через «бутылочное горлышко». Эти результаты подтвердили предположение о том, что разнообразие вируса в Европе и Северной Америке до XX века было значительно больше, чем в поствакцинальный период. Однако они не согласуются с данными о признаках оспенных поражений на мумифицированных останках и наличием древних письменных описаний.
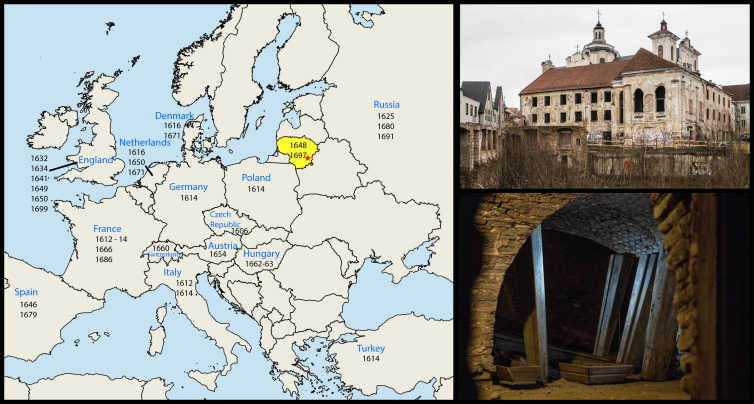 Образец VD21, датированный примерно 1654 годом, найден в доминиканском костеле Святого Духа (Вильнюс, Литва). Черным цветом указаны даты вспышек оспы в соседних странах в XVII в. Внизу справа — склеп, в котором была мумия ребенка (Duggan AT, 2016)
Образец VD21, датированный примерно 1654 годом, найден в доминиканском костеле Святого Духа (Вильнюс, Литва). Черным цветом указаны даты вспышек оспы в соседних странах в XVII в. Внизу справа — склеп, в котором была мумия ребенка (Duggan AT, 2016)
Что помогла выяснить древняя ДНК
Кто исследовал
Итальянские ученые в 2023 году (подробнее на PCR.NEWS).
Какие образцы изучали
Пятьдесят шесть геномов вируса оспы (4 древних, 2 исторических, 48 современных и 2 образца со спорной датировкой), которые общедоступны в GenBank. Древние образцы датированы 603–1000 гг. н.э., и выделены от жителей Северной Европы и западной части России. Эти образцы были обнаружены в результате скрининга данных высокопроизводительного секвенирования скелетных и зубных останков 1867 людей, живших в Евразии и Америке от 150 до 31 630 лет назад.
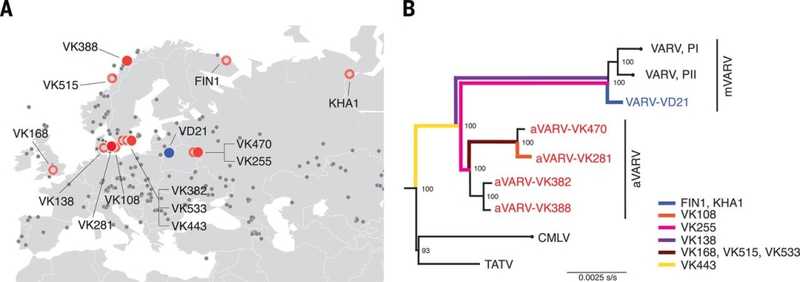 Географическое и филогенетическое расположение образцов вируса оспы. Четыре древних образца отмечены красным. А — карта, показывающая географию обнаружение древней ДНК, В — сокращенное дерево, включающее образцы вирусной ДНК с максимальным качеством (Mühlemann B, 2020)
Географическое и филогенетическое расположение образцов вируса оспы. Четыре древних образца отмечены красным. А — карта, показывающая географию обнаружение древней ДНК, В — сокращенное дерево, включающее образцы вирусной ДНК с максимальным качеством (Mühlemann B, 2020)
Уточненный возраст патогена
Общий MRCA современных и исторических VARV циркулировал 448 лет назад, а только современных — 332 года назад. Это согласуется с полученными ранее результатами. Две современные линии VARV разделились до начала глобальной вакцинации против оспы, после чего прошли через «бутылочное горлышко», вызванное вакцинацией. Сокращение было более экстремальным для линии P-I, чем для линии P-II.
Однако предок всех геномов VARV циркулировал 3,8 тысяч лет назад — то есть примерно на два тысячелетия раньше, чем было показано в работе, посвященной исследованию оспы, поражавшей викингов.
Тогда, в 2020 году международная команда ученых, в которую вошли сотрудники МГУ им. Ломоносова и Музея антропологии и этнографии имени Петра Великого (Кунсткамера) РАН рассчитала, что общий предок вируса оспы существовал 1700 лет назад (подробнее на PCR.NEWS). Геномы эпохи викингов относились к ныне вымершей кладе, сестринской по отношению к современным VARV, циркулировавшей до ликвидации оспы. Эти вирусы имели свой уникальный паттерн инактивации генов. Его анализ позволяет сделать вывод, что они могли заражать более широкий круг хозяев, чем поздние исторические штаммы. И современные, и древние линии вируса оспы были способны вызывать высокую температуру у инфицированных хозяев, так как у них был инактивирован ген B16R, ассоциированный с развитием лихорадки. Но вопрос о том, вызывали ли штаммы VARV эпохи викингов другие проявления заболевания, характерные для исторической оспы, пока остается нерешенным.
Тем не менее данные древней ДНК подтверждают широкое распространение вируса оспы в Древнем мире и примиряют результаты молекулярного датирования с археологическими находками.
Salmonella enterica Paratyphi C, возбудитель паратифа С: «Красота в разнообразии»
Современные варианты появились примерно 339 лет назад.
О патогене
Salmonella enterica — грамотрицательная бактерия, имеющая форму палочки, относится к семейству энтеробактерий. Это повсеместно встречающаяся и стойкая бактерия, которая может выживать до нескольких недель в сухих условиях и нескольких месяцев в воде. Некоторые варианты S. enterica вызывают системную инфекцию и часто ограничиваются одним видом хозяина или имеют узкий круг хозяев. Другие варианты S. enterica вызывают гастроэнтерит различной степени тяжести и поражают широкий круг хозяев.
Классификация
Сегодня 99% случаев сальмонеллеза млекопитающих, в том числе до 200 000 ежегодных смертельных случаев инфекции у человека, связаны с подвидом S. enterica subsp. enterica (S. enterica), который объединяет около 60% всех сероваров. Для человека специфичны серовары Typhi, Paratyphi A и Paratyphi C, вызывающие брюшной тиф и паратифы А и С, соответственно. S. enterica Paratyphi C вызывает системное заболевание преимущественно у людей, но является членом филогенетической линии («линия Para C»), которая также включает серовары, инфицирующие свиней (Typhisuis, Choleraesuis).
Первоначальная гипотеза о происхождении
Кто исследовал
Ученые из США в 1990 году.
Какие образцы изучали
761 изолят сероваров Salmonella Typhi, S. Paratyphi A, S. Paratyphi B, S. Paratyphi C и S. Sendai (адаптированные к человеку агенты кишечной инфекции), а также S. Miami и S. Java (вызывают гастроэнтерит как у людей, так и у животных).
Происхождение
Было высказано предположение, что серовары S. Paratyphi C, S. Choleraesuis и S. Typhisuis произошли от общего предка, который уже был способен вызывать системную инфекцию у свиней. Приобретение антигена Vi позволило серотипу S. Рaratyphi C адаптироваться к человеку. Случаи выявления S. Рaratyphi C у животных позволяют предположить, что адаптация этого варианта к человеку не столь полная, как в случае S. Typhi или S. Paratyphi A. Подтверждает это предположение то, что в некоторых частях мира человек является значимым вторичным хозяином S. Choleraesuis, который вызывает тяжелую кишечную лихорадку с необычно высоким уровнем смертности.
Что помогла выяснить древняя ДНК
- Древняя Евразийская суперветвь
Кто исследовал
Международная команда, включавшая ученых из Казани, Самары, Ставрополя и Москвы, в 2020 году (подробнее на PCR.NEWS).
Какие образцы изучали
Восстановленные геномы восьми штаммов Salmonella enterica, обнаруженные поиском в 2739 древних метагеномах из зубов останков собирателей и скотоводов из Евразии, от России до Турции и Швейцарии. Период захоронений варьировал от неолита до средневековья. Самый древний из геномов S. enterica был обнаружен на территории России (Мурзихинский могильник, Татарстан).
Набор данных для анализа включал 2961 штамм, среди которых, кроме восстановленных, были современные и ранее опубликованные древние геномы. Два таких генома были обнаружены на кладбище, связанном с эпидемией «коколицтли», произошедшей в 1545–1550 годах н. э. в области Тепосколула-Юкундаа на юге Мексики. Один геном был реконструирован из зубов и костей молодой женщины, похороненной около 1200 года в Тронхейме (Норвегия).
Возраст патогена
Все древние геномы сгруппировались в одну филогенетическую ветвь, содержащую ограниченное количество сероваров, которую ученые обозначили как Древняя Евразийская суперветвь (Ancient Eurasian Super Branch, AESB). В ее пределах древние геномы из Мексики и Норвегии стали предковыми для современных штаммов линии Paratyphi C. Геномы раннего бронзового века (Италия, 4200 лет назад) и неолита (Швейцария, 5200 лет назад) сформировали новую ветвь, базальную для всей линии Para C. Другие древние геномы группировались за пределами этой линии. Молекулярное датирование позволило оценить время циркуляции общего предка всех древних и современных представителей линии Para C периодом от 5515 до 2667 лет назад (возраст узла около 4000 лет). Ученые сделали вывод, что древние бактерии S. enterica, исходно вызывающие заболевания домашних животных, с развитием скотоводства и земледелия приобрели адаптацию к новому хозяину — человеку. Получается, что неолитические скотоводы случайно «одомашнили» не только полезных животных, но и сальмонеллу.
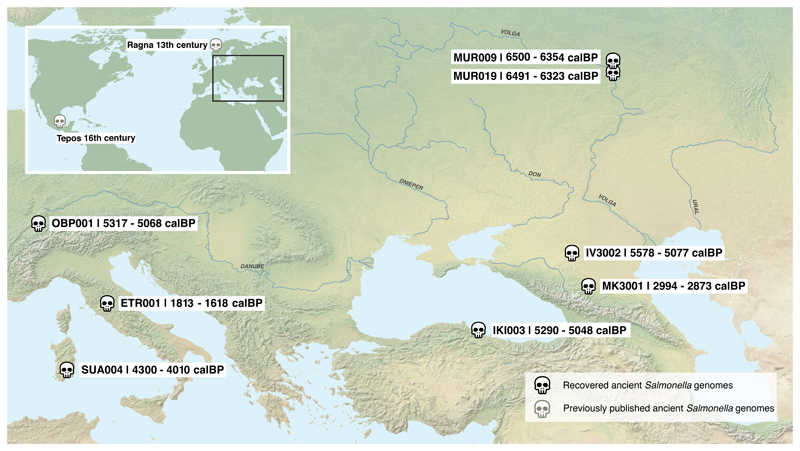 Места обнаружения древних человеческих останков, инфицированных S. enterica, и их возраст на основе радиоуглеродного датирования. В левой верхней части показаны ранее опубликованные древние геномы из Норвегии XIII века и Мексики XVI века (Key FM, 2020)
Места обнаружения древних человеческих останков, инфицированных S. enterica, и их возраст на основе радиоуглеродного датирования. В левой верхней части показаны ранее опубликованные древние геномы из Норвегии XIII века и Мексики XVI века (Key FM, 2020)
Только 60 из 2961 современных геномов S. enterica были частью AESB, в то время как большинство относилось к другим ветвям, для которых не были обнаружены древние представители. Эти результаты позволяют предположить, что обнаруженные доисторические случаи инфекции S. enterica в Евразии были вызваны подгруппой сероваров, в то время гораздо более разнообразной, чем сегодня.
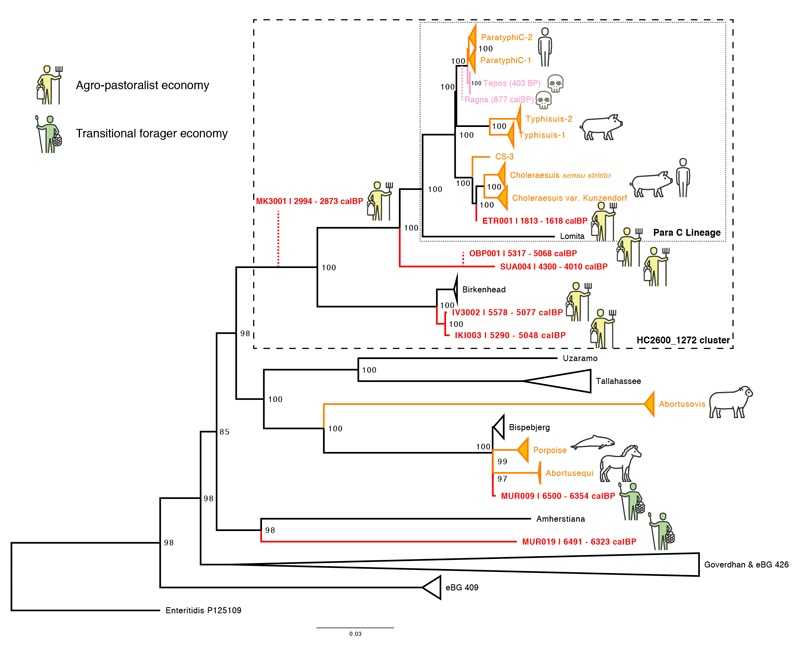 Филогенетические взаимоотношения реконструированных древних и современных геномов S. enterica, относящихся к Древней Евразийской суперветви. Новые древние геномы показаны красным, а ранее опубликованные — розовым. Серовары, адаптированные к хозяину, окрашены в оранжевый цвет (Key FM, 2020)
Филогенетические взаимоотношения реконструированных древних и современных геномов S. enterica, относящихся к Древней Евразийской суперветви. Новые древние геномы показаны красным, а ранее опубликованные — розовым. Серовары, адаптированные к хозяину, окрашены в оранжевый цвет (Key FM, 2020)
- Происхождение линии Para C
Кто исследовал
Команда ученых из Китая и Германии в 2021 году.
Какие образцы изучали
ДНК S. enterica из пульпы зубов шести человек, живших в Синьцзяне (территория современного Китая) в бронзовом веке, около 3000 лет назад. В датасет для анализа вошли четыре ранее опубликованных древних генома и 475 геномов современных штаммов.
Уточненный возраст патогена
На филогенетическом дереве образцы из Синьцзяна группировались как предковые для линии Para C. Время разделения сероваров Paratyphi C, Typhisuis и Choleraesuis в интервале 3088–3219 лет назад (возраст узла примерно 3185 лет назад). Общий предок современных представителей серовара Paratyphi C циркулировал около 339 лет назад.
Treponema pallidum, возбудитель сифилиса: «Колумб не виноват!»
Современные варианты появились примерно 286 лет назад.
О патогене
Бледная трепонема вызывает три вида заболеваний, известные как трепонематозы: сифилис, фрамбезию и беджель. Венерический сифилис распространен во всем мире, каждый год регистрируются миллионы новых случаев. Фрамбезия и беджель чаще всего встречаются в жарких и засушливых условиях — в тропических регионах мира. Все три заболевания передаются при прямом контакте с поражениями кожи или слизистыми оболочками. Сифилис передается половым или врожденным путем, хотя иногда наблюдается случайная передача, например, при переливании крови. Заражение эндемическими трепонематозами (фрамбезией и беджелем) чаще всего происходит через поражения кожи, преимущественно в детском или подростковом возрасте. Все три заболевания имеют сходные клинические проявления, протекают в несколько стадий, включающие повреждение кожи и других тканей.
Классификация
Вид бактерий T. pallidum включает три подвида: T.p. pallidum (TPA), T.p. pertenue (TPE), T.p. endemicum (TEN). TPA вызывает сифилис, TPE — фрамбезию и трепонематоз приматов, TEN — беджель. Филогенетически TPA является монофилетической линией, четко отделяясь от группы TPE/TEN. Внутри этой линии ученые выделили две ветви, обозначенные по названию эталонных штаммов Nichols и SS14. Группа Nichols состоит почти исключительно из образцов, собранных у пациентов в Северной Америке с 1912 по 1986 год. Ветвь SS14 имеет глобальное распространение и включает образцы из Европы, Северной и Южной Америки, собранные с 1951 по 2013 годы.
Первоначальная гипотеза о происхождении
Кто исследовал
Международная команда ученых в 2016 году.
Какие образцы изучали
Семьдесят образцов из 13 стран — 52 мазка на сифилис, взятых от пациентов с 2012 по 2013 годы, и 18 образцов, положительных на сифилис, фрамбезию и беджель, собранных начиная с 1912 года и поддерживаемых на лабораторных кроликах. В анализ вошли сиквенсы высокого качества для 28 из этих штаммов и 11 опубликованных ранее геномов.
Возраст патогена
Используя даты выделения образцов TPA для калибровки, ученые рассчитали среднюю скорость эволюции на уровне 6,6 x 10-7 замен на сайт в год для всего генома, что соответствовало оценкам, полученным для других клональных патогенов человека, таких как Shigella sonnei (6,0 x 10-7) и Vibrio cholerae (8,0 x 10-7). Получилось, что самый ближайший общий предок подвида TPA циркулировал менее 500 лет назад (медианный год 1733, в диапазоне 1588–1848 гг.) — не ранее, чем началась пандемия сифилиса конца XV века.
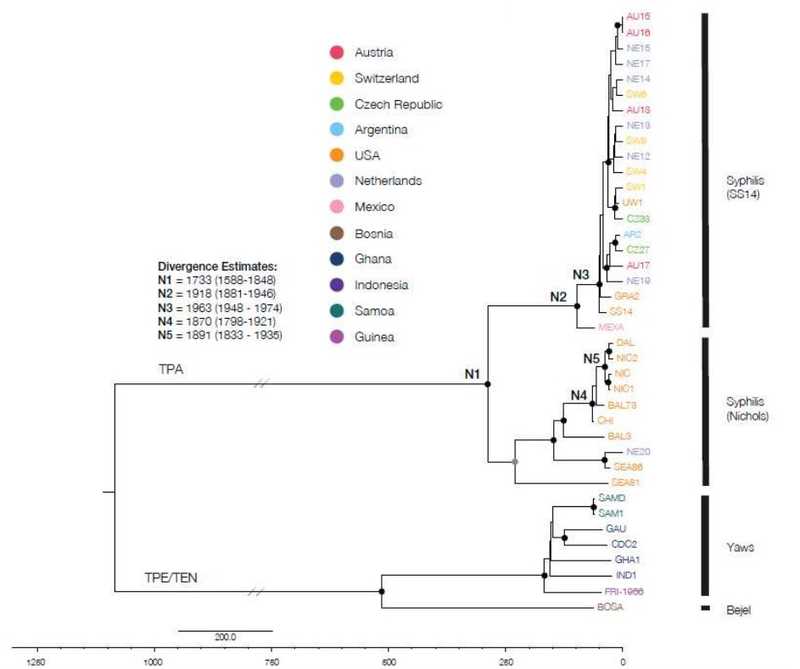 Результаты молекулярного датирования на основе разнообразия современных штаммов T. pallidum (Arora N, 2016)
Результаты молекулярного датирования на основе разнообразия современных штаммов T. pallidum (Arora N, 2016)
Что помогла уточнить древняя ДНК
Кто исследовал
Команда специалистов из Европы в 2020 году и в 2024 году.
Какие образцы изучали
Один образец генома T. pallidum высокого качества возрастом около 1782 лет из ракушечникового кургана Жаботикабейра II у южного побережья Бразилии, в штате Санта-Катарина (подробнее на PCR.NEWS). Четыре древних генома T. pallidum раннего Нового времени, которые ученым удалось реконструировать из человеческих останков человек из часовни Святого Духа в Турку (Финляндия), из погоста в Порвоо (Финляндия), с кладбищ Св. Якоба и Св. Георгия в Тарту (Эстония) и из лазарета Гертруды в Кампене (Нидерланды) (подробнее на PCR.NEWS). В выборку для филогенетического анализа ученые взяли еще 94 последовательности, включая ранее опубликованные геномы TPA и TPE из колониальной Мексики и геномы современных трепонем, выделенных от людей и приматов.
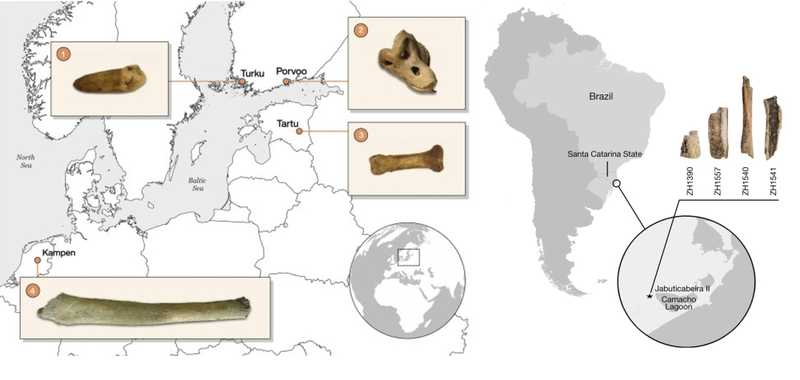 Места обнаружения образцов, для которых удалось получить геномы T. pallidum (адаптировано из [1], [2])
Места обнаружения образцов, для которых удалось получить геномы T. pallidum (адаптировано из [1], [2])
Уточненный возраст патогена
Древний геном из Бразилии занял базальное положение по отношению к современной кладе TEN. Из древних европейских штаммов четыре были наиболее близки к штаммам, вызывающим сифилис, и образовали сестринскую группу для всех TPA. Остальные древние геномы были связаны с ветвью TPE.
Время циркуляции самого недавнего общего предка, рассчитанное для всего семейства T. pallidum, относится к далекой доисторической эпохе — к периоду 12 006–545 гг. до н. э. Общий предок штаммов венерического сифилиса датирован 844 годом н. э, штаммов TPE — 835 годом, а штаммов TEN — 47 годом. MRCA современной группы Nichols циркулировал в 1238 году, а группы SS14 — немного раньше, в 1127 году. Самые молодые штаммы T. pallidum (группа SS14-Ω) возникли около 1738 года.
Из-за включения в анализ древних геномов время расхождения ветвей оказалось более ранним, чем те оценки, которые были сделаны только на основе современных штаммов. Новые временные оценки, обнаружение базальной сестринской линии современных TPA и радиоуглеродная датировка останков указывают на то, что первые случаи заражения сифилисом в Европе произошли до контакта с Новым Светом. Это позволяет предположить, что источник эпидемии конца XV века находился в Старом Свете. Возможно, трепонемные заболевания возникли в Евразии или Африке и попали в Америку вместе с первыми людьми, мигрировавшими туда около 15 тысяч лет назад.
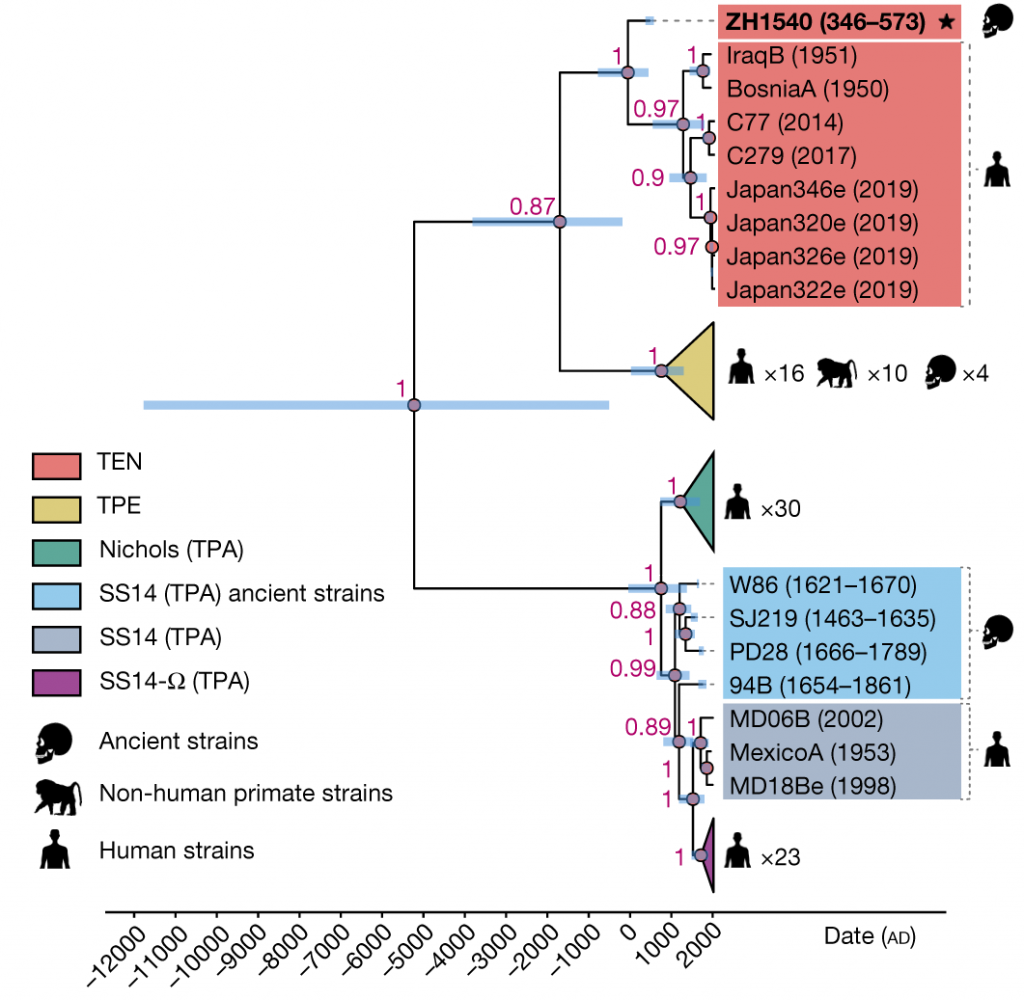 Молекулярное датирование древних и современных геномов T. pallidum (K Majander, 2024)
Молекулярное датирование древних и современных геномов T. pallidum (K Majander, 2024)
В целом скорость и направление эволюционных изменений, биология и взаимоотношения с хозяином уникальны для каждого патогена. Некоторые линии существуют и прекрасно себя чувствуют в человеческой популяции с глубокой древности, а некоторые представляют собой лишь слабые отголоски былого разнообразия. Пока что в области эволюционной истории бактерий и вирусов, связанных с человеком, остается много вопросов. Кропотливая работа ученых помогла пролить свет лишь на некоторые фрагменты, а заполнить оставшиеся пробелы помогут дальнейшие исследования, в особенности анализ древней ДНК, обнаруженной в археологических памятниках.





 0
0