Меню
Меню





 Все темы
Все темы
Пять историй неизлечимых заболеваний
Болезнь Альцгеймера пытаются лечить больше ста лет, бешенство — больше ста никак не лечат. Какие-то болезни сложно понять, на другие — сложно нацелить терапию, третьи вылечить невозможно вовсе, но можно взять под контроль. Рассказываем о пяти неизлечимых болезнях и о надеждах, которые есть сегодня.

Синдром Ретта
Резкий откат развития и последующая инвалидизация, когда ничто не предвещало.
Как это было
В 1983 году в журнале Annals of Neurology вышла статья, в которой шведский педиатр Бенгт Хагберг и его коллеги описали 35 случаев, необычайно похожих друг на друга. Все пациенты — девочки с прогрессирующим поражением мозга — энцефалопатией. Все они первые 7–18 месяцев жизни развивались нормально: осваивали перевороты, учились сидеть и ходить, говорили свои первые слова. Но затем развитие останавливалось — и вскоре обращалось вспять. Девочки становились менее контактными и более беспокойными. За пару лет утрачивали все приобретенные навыки, переставали разговаривать, ходить и даже сидеть. У них появлялись одинаковые стереотипные движения рук, развивалась умственная отсталость и неврологические симптомы — эпилепсия, вазомоторные нарушения, спастики. Затем наступал стационарный период, когда состояние детей не ухудшалось, но и улучшений тоже не было. Никто не знал, в чем дело, однако авторы статьи заметили, что эти симптомы очень похожи на те, которые почти 30 лет назад — в 1954 году — описывал немецкий ученый Андреас Ретт.
 Девочка с синдромом Ретта: характерное положение рук. Wikimedia.org |
CC BY-SA 4.0
Девочка с синдромом Ретта: характерное положение рук. Wikimedia.org |
CC BY-SA 4.0
Ретт тогда обследовал двух девочек с нарушением психического развития и заметил, что они как-то странно — и одинаково — с некоторой периодичностью сжимают руки, как будто моют их. Он вспомнил еще несколько таких случаев из своей практики, а затем поехал по Европе — искать детей с такими же нарушениями. В 1966 году он написал несколько статей в немецкие журналы, но тогда научное сообщество их не заметило. Только после того, как свою статью опубликовал Хагберг, заболевание выделили и назвали синдромом Ретта — по имени первооткрывателя.
Что мы знаем сегодня
Синдром Ретта — генетическое заболевание. Ген, ассоциированный с синдромом Ретта, был открыт только в 1999 году. Это ген MECP2, расположенный на Х-хромосоме, а открыла его американский генетик Худа Зогби. MECP2 кодирует метил-CpG-связывающий белок 2 — в норме этот белок в определенный момент развития выключает несколько других генов, чтобы мозг развивался нормально. Если же в гене мутация и он неактивен, то развитие мозга нарушается.
Синдром Ретта поражает одну из 10–15 тысяч новорожденных, мутация возникает спонтанно. Заболевание встречается только у девочек, потому что Х-хромосомы у них две, и вторая Х-хромосома в порядке. Плод мужского пола с такой мутацией практически всегда погибает еще в утробе. Правда, несколько мальчиков рождались с синдромом Ретта, но у них был еще один синдром — Клайнфейтера, который подарил им лишнюю Х-хромосому.
Тяжесть заболевания у разных пациенток неодинакова, потому что в каждой клетке женского организма работает только одна Х-хромосома — другая инактивируется, превращаясь в тельце Барра. Выключение Х-хромосомы происходит случайным образом, и обычно в половине клеток работает одна, а в другой половине другая. Но соотношение может быть и иным, и чем больше у больной девочки клеток с активной Х-хромосомой без мутаций, тем легче протекает болезнь. Это можно проверить: существует анализ Х-инактивации, который может предсказать течение заболевания.
Помимо описанных в 1984 году симптомов есть и другие: у больных девочек развиваются сколиоз, бруксизм (спазм жевательных мышц и скрежет зубов), мышечная атрофия, нарушения дыхания, проблемы с сердцем. Стационарный период может длиться и до 40 лет, но при этом пациентки не могут сами передвигаться и есть. Смерть обычно наступает из-за остановки сердца или дыхания или от судорог — в очередном приступе эпилепсии.
Какие есть варианты
Лечения нет, но существуют методы реабилитации, позволяющие сохранить старые навыки или приобрести новые. Тем временем исследователи пытаются найти способ лечить болезнь — в основном, конечно, пробуют восстановить нормальную экспрессию гена. Еще в 2007 году ученые из Великобритании обратили вспять неврологические симптомы в мышиной модели синдрома Ретта, восстановив экспрессию деактивированного гена с мутацией. В 2020 году другим ученым тоже удалось активировать MeCP2 у мышей.
В начале этого года ученые из США исправили фенотип синдрома Ретта в эмбриональных стволовых клетках и нейронах in vitro методом эпигеномного редактирования. А в марте FDA одобрило первый препарат для лечения синдрома — трофинетид в виде раствора для приема внутрь Daybue. В двойном слепом плацебо-контролируемом исследовании препарат улучшил движение и поведенческие характеристики у пациенток от 5 до 20 лет. Но как именно действует лекарство — неясно.
Совсем недавно прошел фазу 3 клинических исследований препарат под названием трофинетид — синтетический аналог глицина-пролина-глутамата, N-концевого трипептида белка инсулиноподобного фактора роста 1. Состояние пациенток, получавших препарат, заметно улучшилось.
Болезнь Альцгеймера
Прогрессирующее слабоумие, с которым не могут справиться более ста лет.
Как это было
В 1901 году в больницу для душевнобольных во Франкфурте-на-Майне, где работал психиатр и невролог Алоис Альцгеймер, попала женщина с прогрессирующим слабоумием. Августе Детер был 51 год, и у нее были провалы в памяти, неспособность сконцентрироваться, бессонница, дезориентация, проблемы с речью, письмом и чтением и паранойя: муж, который и привез ее в больницу, говорил, что несколько месяцев назад она стала сильно его ревновать, а временами у нее случались приступы страха и галлюцинации. Под наблюдением врачей женщина находилась еще четыре с половиной года, и все это время ей становилось хуже, в какой-то момент она перестала вставать с кровати. Врачи встречали подобные симптомы и раньше, но обычно ими страдали люди постарше.
Пациентка умерла в 56 лет от сепсиса. Альцгеймер, уже несколько лет не работающий в той больнице, попросил прислать ему образцы тканей ее мозга. В микроскоп он увидел потерю нейронов и сразу два типа отложений — сенильные бляшки вокруг нейронов (такие уже наблюдали исследователи в мозге больных старческой деменцией и эпилепсией) и нейрофибриллярные клубки — внутри.
В 1910 году коллега Альцгеймера Эмиль Крепелин описал деменцию с ранним началом и бляшками как болезнь Альцгеймера — и название закрепилось. Он же назвал ее пресенильной, то есть «предстарческой» деменцией. И долгое время болезнь Альцгеймера диагностировали пациентам 45–65 лет. Но в 1977 году на конференции, посвященной болезни, исследователи сошлись на том, что и сенильная, старческая деменция проявляется примерно так же. Со временем заболевание стали диагностировать и людям старше 65 лет.
 Credit: 123rf.com
Credit: 123rf.com
Что мы знаем сегодня
Бляшки в нейронах оказались нерастворимыми агрегатами бета-амилоида Aβ, а нейрофибриллярные клубки — скоплениями гиперфосфорилированного тау-белка. Именно они по сей день считаются главными биомаркерами болезни Альцгеймера.
Бета-амилоиды — небольшие пептиды примерно из 40 аминокислот — получаются при расщеплении мембранного белка-предшественника бета-амилоида. Сами по себе они даже полезны: стимулируют нейрогенез. Но структура этих пептидов такова, что им очень легко прилипнуть друг к другу, и такие агрегаты из нескольких пептидов уже становятся опасными, они вызывают образование активных форм кислорода, воспаление и гибель нейронов.
Тау-белок входит в состав микротрубочек, которые образуют «скелет» нейронов и их длинных отростков — аксонов. К такому белку могут цепляться фосфатные группы, и, если фосфатов отчего-то присоединится слишком много, он отделится от микротрубочек и начнет образовывать нейротоксичные клубки с такими же белками.
Однако амилоидные бляшки обнаруживают и у здоровых людей, а у некоторых пациентов с болезнью Альцгеймера таких бляшек мало. А вот количество агрегатов тау-белка обычно коррелирует с тяжестью нейродегенерации. Основная на сегодня гипотеза амилоидного каскада тем временем гласит, что именно патологические формы бета-амилоида приводят к гиперфосфорилированию тау-белка и вообще запускают развитие болезни.
С момента постановки диагноза до смерти обычно проходит 7–10 лет. Постепенно пациент теряет все больше навыков и в конце концов становится недееспособным.
А раннее начало деменции сегодня связывают с генетической предрасположенностью. Причиной может быть мутация в одном из генов белка пресенилина, или в гене белка-предшественника бета-амилоида (APP), или еще в каком-нибудь гене. Тогда первые симптомы проявляются уже в 44 года, а к 50 годам пациент теряет рассудок. Такие больные умирают от осложнений примерно в 60 лет. Возможно, Августа Детер как раз была носительницей такой мутации.
Есть и хорошая новость: от наследственной формы болезни Альцгеймера могут защитить другие мутации, хоть это и редкость. На сегодняшний день известно о двух таких пациентах — мутации в генах APOE3 (вариант R136S, или Крайстчерч) и RELN (H3447R, или вариант RELN-COLBOS) отсрочили деменцию почти до 70 лет.
Какие есть варианты
Работающего лечения болезни Альцгеймера пока не придумали. Хотя несколько препаратов одобрены по всему миру, они ничего не лечат.
Первыми одобрили три ингибитора холинэстеразы, которые должны улучшать передачу импульсов между нейронами. Подобные препараты используют и для лечения других нейродегенеративных заболеваний, но они лишь слегка ослабляют симптомы. В 2003 году зеленый свет дали регулятору глутаматных рецепторов: при болезни Альцгеймера работа таких рецепторов нарушается. Однако и в его эффективности есть сомнения. Позднее на арену вышли принципиально новые лекарства — препараты на основе моноклональных антител к бета-амилоиду. Их стали производить все, кто мог.
Один из таких препаратов — адуканумаб — FDA одобрило в 2021 году. А в начале этого года в ускоренном порядке было одобрено еще одно аналогичное лекарство — леканемаб; оба препарата выпустила на рынок компания Biogen. Однако же счастливых пациентов больше не стало, а вот вопросов к новым лекарствам прибавилось. Количество амилоидных бляшек в мозгах пациентов они снижают, и когнитивный спад иногда происходит немного медленнее. Однако неясно, насколько они безопасны: в клинических испытаниях третьей фазы препарат привел к инфузионным реакциям у 26,4% участников, а у 29,9% — к аномалиям, связанным с амилоидами ( ARIA), включая отеки мозга (ARIA-E, 12,6%) и мозговые кровоизлияния (ARIA-H, 17,3%). В группе плацебо аномалии, связанные с амилоидами, встречались в 10,7% случаев. Три пациента умерло (1, 2) во время испытаний леканемаба. При этом препарат очень дорогой — год лечения леканемабом обойдется более чем в 20–25 тысяч долларов (дозировка рассчитывается по весу). Тем не менее, в июле этого года FDA одобрило леканемаб полностью.
Ученые постоянно ищут новые способы снизить количество отложений бета-амилоида и тау-белка в мозге и замедлить нейродегенерацию (а некоторые даже пытаются улучшить состояние пациентов или модельных лабораторных животных). Для этого они увеличивают экспрессию защитных белков в мозге мышей, разрушают ферменты, редактируют гены и даже пытаются влиять на бета-амилоиды через сон (1, 2).
Бешенство
Смертельная инфекция, которая поражает нервную систему и убивает за считанные дни.
Как это было
Первым упоминаниям о смертельной болезни, которая поражает диких и домашних животных и делает их буйными и агрессивными, несколько тысяч лет. В I веке н. э. Корнелий Цельс описал подобную человеческую болезнь, которой можно заразиться после укуса собаки. Он назвал ее водобоязнью, потому что неспособность глотать воду и любые другие жидкости — один из основных ее симптомов.
А в 1884 году два французских микробиолога взяли умершего от бешенства кролика, вынули его спинной мозг и две недели высушивали его в колбе. Затем то, что осталось от кроличьего мозга, они ввели в мозг зараженной бешенством собаки — и та не умерла. Так Луи Пастер и Эмиль Ру, не знавшие еще ничего о вирусах, создали вакцину от бешенства.
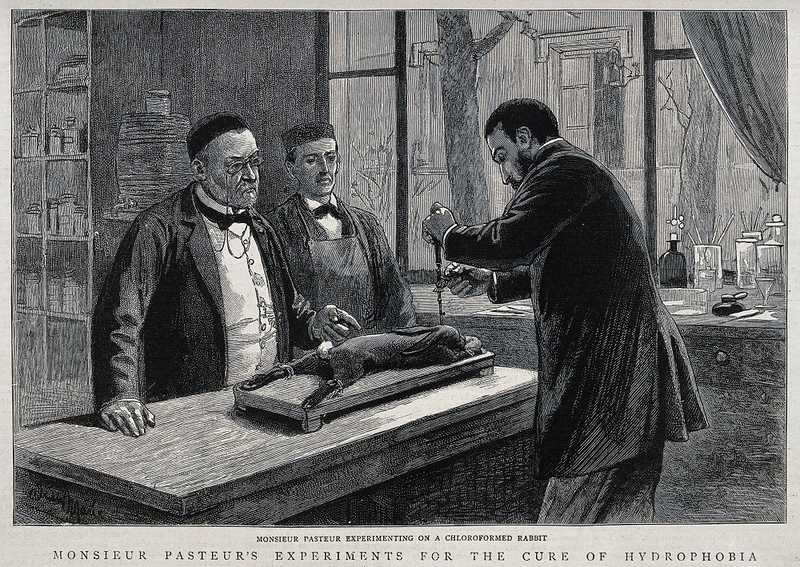 Луи Пастер (слева) наблюдает, как его коллега трепанирует кролика, усыпленного хлороформом, во время исследования вакцины против бешенства. 1885
Луи Пастер (слева) наблюдает, как его коллега трепанирует кролика, усыпленного хлороформом, во время исследования вакцины против бешенства. 1885
Все было, конечно, не так просто. Пастеру и его коллегам понадобилось почти пять лет, чтобы научиться ослаблять вирус бешенства и иммунизировать им собак и кроликов. За это время погибло очень много животных, но антирабическую вакцину — вакцину от вируса бешенства — создать удалось. Оставалось проверить ее на людях.
Рисковать было страшно. Однако во Франции тогда то и дело случались вспышки бешенства, а с зараженными людьми жестоко расправлялись. И однажды к Пастеру обратилась мать девятилетнего Йозефа Майстера, искусанного бешеной собакой: мальчик спас от нее несколько человек, но пострадал сам. Смерть была вопросом времени. За две недели ему сделали 14 прививок высушенного мозга кролика, вводя каждый раз все более свежий образец, а значит — более вирулентный. Спустя месяц после последней дозы мальчик был здоров — бешенством он не заболел. С тех пор от бешенства стали прививать и других людей.
Что мы знаем сегодня
Бешенство вызывает РНК-вирус Neuroiyctes rabid из семейства рабдовирусов (Rhabdovtridae). Выглядит он как пуля размером 100–200 нанометров. Этот вирус не один — известно семь его генотипов. Вирус поражает центральную нервную систему, роговицу глаз и слюнные железы. Через слюну он и передается — при укусе или любой другой контакт зараженной слюны и слизистых. А водобоязнь больных — на самом деле спазмы глотки и гортани, не дающие больному эту слюну проглотить (и любую другую жидкость — тоже). Так у вируса больше шансов найти новых хозяев: больные агрессивны, разбрызгивают слюну вокруг себя, да еще и могут кусаться.
Вирус выдерживает замораживание, но разрушается под действием прямых солнечных лучей, кислот, щелочей и этанола, поэтому после укуса рану прежде всего нужно тщательно промыть с мылом и обработать чем-то спиртосодержащим. И уже затем — обращаться к врачу.
Ежегодно в мире заболевает более 50 тысяч человек, главным образом в Юго-Восточной Азии. Диагностировать бешенство лабораторно до появления симптомов невозможно.
Какие есть варианты
Вариант и сто лет назад, и сегодня только один — вакцинация. Правда, вакцина способна помочь только до появления первых симптомов. У зараженного есть от 10 до 90 дней после контакта с предположительно бешеным животным, чтобы поставить прививку, пока вирус не добрался до нервной системы. И хотя риск заболеть различается в зависимости от места укуса (чем ближе к голове, тем риск выше), он есть, даже если животное просто облизало царапину на руке или ноге. После окончания инкубационного периода болезнь остановить почти невозможно, и, скорее всего, человек умрет в течение 7–10 дней.
Вначале возникнет головокружение, тошнота, беспокойство и страх, спустя два-три дня появится водобоязнь, судороги глотки и гортани, возбуждение, агрессия, возможно, галлюцинации. Спустя еще несколько дней наступит паралич дыхательной или сердечной мускулатуры — и смерть.
Фибродисплазия оссифицирующая прогрессирующая
Костная ткань появляется там, где ее не должно быть, — на месте мышц, связок и сухожилий.
Как это было
О женщине, которая «стала твердой, как дерево» упоминал французский врач Гай Пэтин в 1692 году в письме. В 1736 году к Джону Фреку, британскому хирургу, попал подросток с шишками по всей спине. Тогда врач назвал болезнь миозитом оссифицирующим прогрессирующим, отразив в этом названии то, что увидел: мышцы превращаются в кости.
Спустя 250 лет, в 1970-х, миозит в названии поменяли на фибродисплазию: тогда американский врач Виктор Маккьюзик отметил, что костная ткань замещает не только мышцы, но и другие ткани — сухожилия и связки.
В конце 1980-х американские врачи Фредерик Каплан и Майкл Заслофф запустили исследовательский проект по изучению ФОП на базе медицинского университета Пенсильвании, а затем там открылась молекулярная лаборатория.
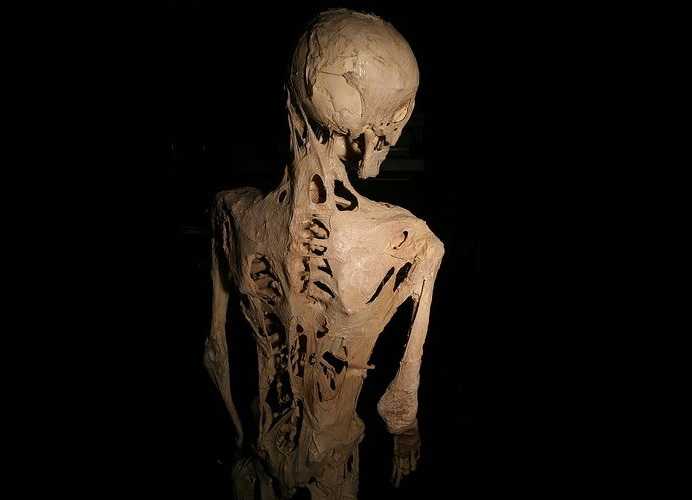 Скелет Гарри Рэймонда Истлака (1933–1973), одного из самых известных пациентов с прогрессирующей оссифицирующей фибродисплазиией. Wikimedia.org
CC BY-SA 3.0
Скелет Гарри Рэймонда Истлака (1933–1973), одного из самых известных пациентов с прогрессирующей оссифицирующей фибродисплазиией. Wikimedia.org
CC BY-SA 3.0
Что мы знаем сегодня
В 2006 году в той самой лаборатории Заслофф и Каплан с коллегами выяснили, что причина болезни — мутация в гене ACVR1, который расположен на второй хромосоме. Ген кодирует рецептор активина А типа I. Такие рецепторы вместе с ACVR-2, вторым типов активиновых рецепторов, расположены в мембране клеток и могут распознавать костные морфогенетические белки (BMP) или активин А — так в нужный момент стволовые клетки получают сигнал дифференцироваться в клетки костной ткани. При ФОП рецептор работает неправильно и стволовые стромальные клетки в мышцах, сухожилиях и связках активируются спонтанно либо после травмы.
Спровоцировать рост костной ткани там, где ее не должно быть, может любое повреждение — даже укол шприца. Как следствие, оперировать бесполезно: проблема лишь усугубится. Вероятно, в окостенении тканей участвует еще иммунная система, однако весь молекулярный механизм неизвестен. Помимо образования лишних костей, у пациентов нарушается регенерация мышц.
Заболевание очень редкое: обычно мутация возникает у ребенка случайно, и таких случаев 1,36 на миллион. Также мутация наследуется по аутосомно-доминантному принципу, то есть можно получить ее от больного родителя с вероятностью 50%.
У детей с ФОП большие пальцы ног часто укорочены и завернуты внутрь — это верный признак болезни; бывают и другие деформации пальцев. Однако диагностика затруднена по сей день: может пройти до семи лет, прежде чем пациенту с ФОП поставят диагноз. Ранние проявления фибродисплазии у детей часто путают с онкологией, потому что выглядят они как подкожные уплотнения на шее, спине или руках. Неверные диагнозы и вмешательства ожидаемо только вредят.
К двадцати годам больные ФОП обычно уже лежат и совсем не могут двигаться — даже рот не открывается. Непораженными остаются гладкая мускулатура, сердце, диафрагма, мышцы глаз, языка и губ. Дольше 40–50 лет пациенты живут редко: в какой-то момент у них развивается дыхательная недостаточность из-за деформаций грудной клетки, кости которой больше не могут нормально двигаться.
Какие есть варианты
Лечения от этой болезни тоже пока не придумали. Иногда используют нестероидные противовоспалительные или гормональные препараты, чтобы предотвратить воспаление и кальцификацию тканей.
Найти лекарство пытаются. Около десяти лет назад канадская фармкомпания (ее уже купила американская компания Ipsen) разработала препарат паловаротен (palovarotene). Он стимулирует рецепторы ретиноевой кислоты и должен мешать появлению новых очагов окостенения. Недавно препарат испытали на пациентах — он вроде бы уменьшил образование оссификатов, однако в 3,3 раза повысил риск переломов позвоночника у детей и снизил минеральную плотность костей. А у 36,8% пациентов младше 14 лет наблюдалось преждевременное закрытие зон роста костей (эпифизов). В январе 2022 года в Канаде лекарство одобрили. Однако американский регулятор FDA и Европейский комитет по лекарственным средствам с одобрением не спешат.
Рассеянный склероз
Человек становится инвалидом из-за того, что оболочки его нервных волокон разрушаются клетками его же иммунной системы.
Как это было
В 1433 году 15-летняя девочка из Южной Голландии, катаясь на коньках, упала и сломала ребро. Через какое-то время после этого ей стало сложно ходить, появились головные и зубные боли, стало нарушаться зрение. А к 19 годам у нее были парализованы обе ноги. Она прожила до 53 лет, и, согласно записям биографов, большую часть жизни была почти полностью парализована — кроме левой руки. Лидвину из Схидама знали как целительницу и еще при жизни называли святой. Сейчас католики почитают ее как покровительницу больных.
А в 1868 году психиатр Жан-Мари Шарко разглядел в мозге умершей пациентки, которая страдала тремором и невнятной речью, хаотично разбросанные участки соединительной ткани на месте нервной — очаги склероза. Он и назвал болезнь рассеянным склерозом и описал триаду симптомов, которые наблюдал у таких пациентов: нистагм (подергивания глазных яблок), интенционный тремор (дрожание конечностей во время целенаправленного движения) и скандированную речь — человек говорит медленно, отрывочно, по слогам.
Только в 1916 году патологоанатом Джеймс Доусон увидел в микроскоп разрушение миелиновых оболочек отростков нейронов — причину всех симптомов, от нарушения зрения до паралича. В 1935 году Томас Риверс показал, что заболевание аутоиммунное: ввел мышам основной белок миелина, чтобы вызвать похожую реакцию иммунитета и симптоматику. К 1960 году стало понятно, что в рассеянном склерозе участвуют Т- и В-лимфоциты.
Некоторые ученые полагают, что святая Лидвина из Схидама тоже болела рассеянным склерозом. А падение могло быть первым симптомом: больные страдают головокружениями.
Что мы знаем сегодня
Рассеянный склероз — аутоиммунное заболевание, которое проявляется довольно рано — в среднем в 30 лет. Болеют чаще женщины. Клетки иммунной системы разрушают миелин, который покрывает аксоны — длинные отростки нейронов. В итоге нарушается передача сигналов.
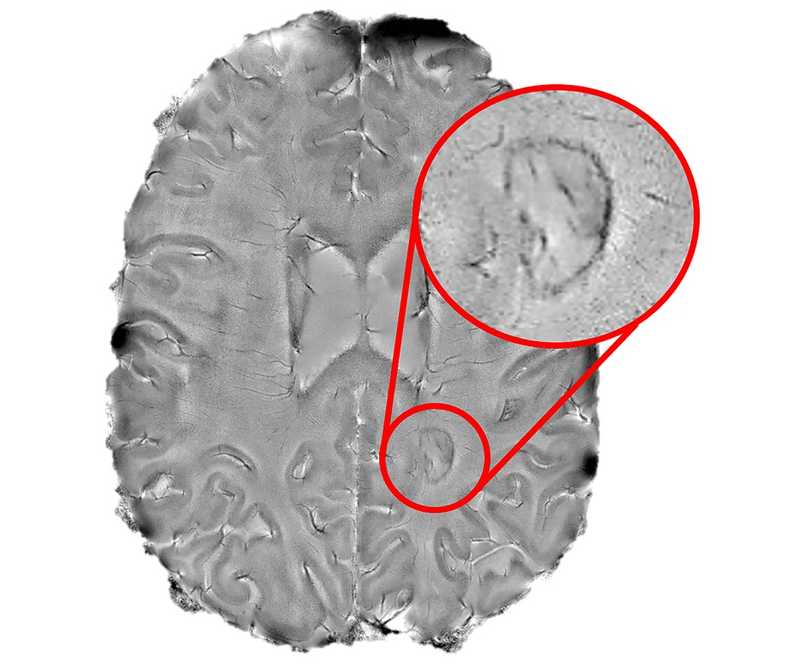 Пятно с темной каймой в очаге хронического воспаления может быть отличительной чертой более агрессивных, инвалидизирующих форм рассеянного склероза (данные МРТ). NIH Image Gallery | J Clin Invest. 2016. DOI:
10.1172/JCI86198
Пятно с темной каймой в очаге хронического воспаления может быть отличительной чертой более агрессивных, инвалидизирующих форм рассеянного склероза (данные МРТ). NIH Image Gallery | J Clin Invest. 2016. DOI:
10.1172/JCI86198
Первыми обычно поражаются миелиновые оболочки зрительных нервов, человеку становится больно двигать глазами, портится зрение, в глазах может двоиться. Из-за поражений мозжечка нарушается координация, возникает тремор. Еще нарушается чувствительность, появляются онемение, боли и парестезии. Бывают нарушения подвижности вплоть до паралича, проблемы с мочеиспусканием или недержание мочи, сексуальная дисфункция.
Болезнь диагностируют примерно у 2–4 человек из 10000. Различают регионы высокого и низкого риска — чем дальше от экватора живет человек, тем риск выше, и наоборот. Возможно, нехватка витамина D способна спровоцировать болезнь. Еще триггером бывает инфекция (болезнь связывают с вирусом Эпштейна — Барр, который поражает В-лимфоциты) и даже ретровирусы — часть нашего собственного генома. Гены, особенно связанные с иммунитетом, тоже играют роль, хотя одного конкретного гена, ответственного за болезнь, нет. И до конца не ясно, почему иммунная система в каких-то случаях вдруг начинает атаковать клетки собственного организма, а в других аналогичных — нет.
Рассеянный склероз чаще протекает с обострениями и периодами покоя (ремиттирующая форма), но у некоторых пациентов развивается непрерывно (прогрессирующая форма). Нередко первая форма со временем — к 40–50 годам — перетекает во вторую, а иногда при прогрессирующей форме случаются еще более сильные обострения.
Какие есть варианты
Можно облегчить течение болезни и снизить число обострений, а значит — отсрочить тяжелые последствия.
Диагностируют болезнь с помощью МРТ или исследования вызванных потенциалов. Нарушение вызванных потенциалов заметно раньше, чем на МРТ становятся видны очаги. Далее — назначают препараты, изменяющие течение болезни (ПИТРС). Это гормональные препараты, снижающие воспаление, иммуномодуляторы (интерфероны, глатирамера ацетат, моноклональные антитела к В-клеткам), снижающие активность иммунитета, иногда используют плазмаферез.
Еще применяют химиотерапию — разрушают иммунные клетки, а потом пересаживают пациенту его же гемопоэтические стволовые клетки, и иммунитет становится толерантным к миелину.
В последние годы ученые пробуют создать мРНК-вакцину от рассеянного склероза. На мышах она уже сработала.




 0
0